Table of Contents
Cover
Title page
Copyright page
PREFACE
ACKNOWLEDGMENTS
PART I: SYNTHETIC TOOLBOX 1: RETROSYNTHESIS AND PROTECTIVE GROUPS
CHAPTER 1.1 RETROSYNTHETIC ANALYSIS
RETROSYNTHESIS BY FUNCTIONAL GROUP INTERCONVERSION (FGI)
RETROSYNTHESIS BY MAKING A DISCONNECTION
CHAPTER 1.2 PROTECTIVE GROUPS
PROTECTION OF KETONES AND ALDEHYDES
PROTECTION OF ALCOHOLS
PROTECTION OF CARBOXYLIC ACIDS
PROTECTION OF AMINES
PART I PROBLEMS: PROTECTIVE GROUPS
PART II: SYNTHETIC TOOLBOX 2: OVERVIEW OF ORGANIC TRANSFORMATIONS
CHAPTER 2.1 NUCLEOPHILES AND ELECTROPHILES
COMMON NUCLEOPHILES
COMMON ELECTROPHILES
CHAPTER 2.2 OXIDATION AND REDUCTION REACTIONS
OVERVIEW OF OXIDATIONS AND REDUCTIONS
COMMON OXIDATION REACTIONS AND OXIDIZING AGENTS
COMMON REDUCTION REACTIONS AND REDUCING AGENTS
PART II PROBLEMS: NUCLEOPHILES, ELECTROPHILES, AND REDOX
PART III: SYNTHESIS OF MONOFUNCTIONAL TARGET MOLECULES (1-FG TMs)
CHAPTER 3.1 SYNTHESIS OF ALCOHOLS (ROH)
SYNTHESIS OF ALCOHOLS BY FUNCTIONAL GROUP INTERCONVERSION (FGI)
SYNTHESIS OF ALCOHOLS BY THE GRIGNARD REACTION
SYNTHESIS OF PROPARGYLIC ALCOHOLS (RC≡CCH2OH)
SYNTHESIS OF PHENOL DERIVATIVES (ArOH)
EXAMPLE: ALCOHOL TM
CHAPTER 3.2 SYNTHESIS OF ALKYL (RX) AND ARYL HALIDES (ArX)
SYNTHESIS OF ALKYL HALIDES VIA FREE-RADICAL HALOGENATION
SYNTHESIS OF ALKYL HALIDES VIA FGI
RETROSYNTHESIS OF ALKYL HALIDES
SYNTHESIS OF ARYL HALIDES (ArX)
EXAMPLE: ALKYL HALIDE TM
CHAPTER 3.3 SYNTHESIS OF ETHERS (ROR′)
WILLIAMSON ETHER SYNTHESIS (RX + R′O− → ROR′)
ALTERNATE ETHER PREPARATIONS
SYNTHESIS OF EPOXIDES
RETROSYNTHESIS OF ETHERS
EXAMPLE: ETHER TM
CHAPTER 3.4 SYNTHESIS OF THIOLS (RSH) AND THIOETHERS (RSR′)
EXAMPLE: THIOETHER TM
CHAPTER 3.5 SYNTHESIS OF AMINES (RNH2) AND ANILINES (ArNH2)
SYNTHETIC EQUIVALENTS OF NH3 (RX → RNH2)
SYNTHESIS OF AMINES VIA REDUCTION REACTIONS
RETROSYNTHESIS OF AMINES
SYNTHESIS OF ANILINE DERIVATIVES (ArNH2)
EXAMPLE: AMINE TM
CHAPTER 3.6 SYNTHESIS OF ALKENES (R2C=CR2)
SYNTHESIS OF ALKENES VIA FGI
SYNTHESIS OF ALKENES VIA THE WITTIG REACTION
EXAMPLE: ALKENE TM
CHAPTER 3.7 SYNTHESIS OF ALKYNES (RC≡CR′)
SYNTHESIS OF ALKYNES VIA FGI
SYNTHESIS OF ALKYNES FROM OTHER ALKYNES (RC≡CH → RC≡CR′)
EXAMPLE 1: ALKYNE TM
EXAMPLE 2: ALKYNE TM
CHAPTER 3.8 SYNTHESIS OF ALKANES (RH)
SYNTHESIS OF ALKANES VIA FGI
SYNTHESIS OF ALKANES VIA C−C BOND FORMATION
RETROSYNTHESIS OF ALKANES
EXAMPLE: ALKANE TM
CHAPTER 3.9 SYNTHESIS OF ALDEHYDES AND KETONES (RCHO, R2C=O)
SYNTHESIS OF ALDEHYDES/KETONES VIA FGI
SYNTHESIS OF ALDEHYDES/KETONES VIA ACYL SUBSTITUTIONS
SYNTHESIS OF KETONES VIA α-ALKYLATION
EXAMPLE: KETONE TM
CHAPTER 3.10 SYNTHESIS OF CARBOXYLIC ACIDS (RCO2H)
SYNTHESIS OF CARBOXYLIC ACIDS VIA FGI
SYNTHESIS OF CARBOXYLIC ACIDS VIA GRIGNARD (RMgBr + CO2 → RCO2H)
CARBOXYLIC ACIDS VIA α-ALKYLATION: MALONIC ESTER SYNTHESIS
EXAMPLE: CARBOXYLIC ACID TM
CHAPTER 3.11 SYNTHESIS OF CARBOXYLIC ACID DERIVATIVES
RELATIVE REACTIVITIES OF CARBOXYLIC ACID DERIVATIVES (RCOLG)
SYNTHESIS OF ACID CHLORIDES (RCOCl)
SYNTHESIS OF ANHYDRIDES (RCO2COR)
SYNTHESIS OF ESTERS (RCO2R)
SYNTHESIS OF AMIDES (RCONH2)
SYNTHESIS OF NITRILES (RC≡N)
EXAMPLE: CARBOXYLIC ACID DERIVATIVE
PART III PROBLEMS: MONOFUNCTIONAL TARGET MOLECULES (1-FG TMs)
PART IV: SYNTHESIS OF TARGET MOLECULES WITH TWO FUNCTIONAL GROUPS (2-FG TMs)
CHAPTER 4.1 SYNTHESIS OF β-HYDROXY CARBONYLS AND α,β-UNSATURATED CARBONYLS
THE ALDOL REACTION
SYNTHESIS OF α,β-UNSATURATED ESTERS USING THE WITTIG REACTION
EXAMPLE: α,β-UNSATURATED CARBONYL
CHAPTER 4.2 MORE ENOLATE REACTIONS: SYNTHESIS OF 1,3-DICARBONYLS, 1,5-DICARBONYLS, AND CYCLOHEXENONES
THE CLAISEN CONDENSATION
THE MICHAEL REACTION
SUMMARY OF ENOLATE SYNTHESES
ROBINSON ANNULATION
CHAPTER 4.3 “ILLOGICAL” DISCONNECTIONS: UMPOLUNG (POLARITY REVERSAL)
SYNTHESIS OF TMs WITH A 1,2-DIOXYGENATED PATTERN
SYNTHESIS OF TMs WITH A 1,4-DIOXYGENATED PATTERN
SYNTHESIS OF TMs WITH A 1,6-DICARBONYL PATTERN
PART IV PROBLEMS: TARGET MOLECULES WITH TWO FUNCTIONAL GROUPS (2-FG TMs)
PART V: SYNTHESIS OF AROMATIC TARGET MOLECULES
CHAPTER 5.1 ELECTROPHILIC AROMATIC SUBSTITUTION (ArH + E+ → ArE)
MECHANISM OF THE ELECTROPHILIC AROMATIC SUBSTITUTION
ELECTROPHILES FOR THE ELECTROPHILIC AROMATIC SUBSTITUTION
ELECTROPHILIC AROMATIC SUBSTITUTION ON SUBSTITUTED BENZENES
RETROSYNTHESIS OF AROMATIC TMs (ELECTROPHILIC AROMATIC SUBSTITUTION)
EXAMPLE: AROMATIC TM 1
CHAPTER 5.2 SYNTHESIS OF AROMATIC TMs VIA DIAZONIUM SALTS (ArN2+ + Nu → ArNu)
PREPARATION OF DIAZONIUM SALTS (ArNH2 → ArN2+)
USE OF DIAZONIUM SALTS (ArN2+ + Nu → ArNu)
RETROSYNTHESIS OF AROMATIC TMs (VIA DIAZONIUM SALTS)
EXAMPLE: AROMATIC TM 2
CHAPTER 5.3 NUCLEOPHILIC AROMATIC SUBSTITUTION (ArX + Nu: → ArNu)
MECHANISM OF NUCLEOPHILIC AROMATIC SUBSTITUTION (SNAr)
RETROSYNTHESIS OF AROMATIC TMs (SNAr)
EXAMPLE: AROMATIC TM 3
PART V PROBLEMS: AROMATIC TMs
PART VI: SYNTHESIS OF COMPOUNDS CONTAINING RINGS
CHAPTER 6.1 SYNTHESIS OF CYCLOPROPANES
RETROSYNTHESIS OF CYCLOPROPANE TMs
CHAPTER 6.2 SYNTHESIS OF CYCLOBUTANES
RETROSYNTHESIS OF CYCLOBUTANE TMs
CHAPTER 6.3 SYNTHESIS OF FIVE-MEMBERED RINGS (RADICAL CYCLIZATION REACTIONS)
BALDWIN’S RULES
RETROSYNTHESIS OF METHYLCYCLOPENTANE TMs
EXAMPLE: METHYLCYCLOPENTANE TM
CHAPTER 6.4 SYNTHESIS OF SIX-MEMBERED RINGS (DIELS–ALDER REACTION)
THE DIENOPHILE (E+)
THE DIENE (Nu:)
REGIOCHEMISTRY OF THE DIELS–ALDER
RETROSYNTHESIS OF CYCLOHEXENES (DIELS–ALDER)
RETROSYNTHESIS OF 1,4-CYCLOHEXADIENES
PART VI PROBLEMS: CYCLIC TMs
PART VII: PREDICTING AND CONTROLLING STEREOCHEMISTRY
CHAPTER 7.1 REACTIONS THAT FORM RACEMATES
FORMATION OF NEW CHIRAL CARBONS
LOSS OF A GROUP FROM A CHIRAL CARBON: RACEMIZATION
CHAPTER 7.2 SN2 MECHANISM: BACKSIDE ATTACK
CHAPTER 7.3 ELIMINATION MECHANISMS
COPE ELIMINATION (SYN)
CHAPTER 7.4 ADDITIONS TO ALKENES AND ALKYNES
SYN ADDITIONS
ANTI ADDITIONS
CHAPTER 7.5 ADDITIONS TO CARBONYLS
DIASTEREOSELECTIVITY IN ACYCLIC SYSTEMS: CRAM’S RULE, FELKIN–AHN MODEL
CHELATION CONTROL BY NEIGHBORING GROUPS
ADDITION TO CYCLOHEXANONES
CHAPTER 7.6 ADDITIONS TO ENOLATES: ALDOL STEREOCHEMISTRY
FORMATION OF (E) AND (Z) ENOLATES
ALDOL REACTION WITH (E) AND (Z) ENOLATES
EXAMPLES: PREDICTING ALDOL STEREOCHEMISTRY
CHAPTER 7.7 ENANTIOSELECTIVITY AND ASYMMETRIC SYNTHESES
PROCHIRAL ENVIRONMENTS
ENANTIOSELECTIVE TECHNIQUES
PART VII PROBLEMS: STEREOCHEMISTRY
SOLUTIONS TO PROBLEMS
Index
Copyright © 2012 by John Wiley & Sons, Inc. All rights reserved.
Published by John Wiley & Sons, Inc., Hoboken, New Jersey.
Published simultaneously in Canada.
No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, scanning, or otherwise, except as permitted under Section 107 or 108 of the 1976 United States Copyright Act, without either the prior written permission of the Publisher, or authorization through payment of the appropriate per-copy fee to the Copyright Clearance Center, Inc., 222 Rosewood Drive, Danvers, MA 01923, (978) 750-8400, fax (978) 750-4470, or on the web at www.copyright.com. Requests to the Publisher for permission should be addressed to the Permissions Department, John Wiley & Sons, Inc., 111 River Street, Hoboken, NJ 07030, (201) 748-6011, fax (201) 748-6008, or online at http://www.wiley.com/go/permissions.
Limit of Liability/Disclaimer of Warranty: While the publisher and author have used their best efforts in preparing this book, they make no representations or warranties with respect to the accuracy or completeness of the contents of this book and specifically disclaim any implied warranties of merchantability or fitness for a particular purpose. No warranty may be created or extended by sales representatives or written sales materials. The advice and strategies contained herein may not be suitable for your situation. You should consult with a professional where appropriate. Neither the publisher nor author shall be liable for any loss of profit or any other commercial damages, including but not limited to special, incidental, consequential, or other damages.
For general information on our other products and services or for technical support, please contact our Customer Care Department within the United States at (800) 762-2974, outside the United States at (317) 572-3993 or fax (317) 572-4002.
Wiley also publishes its books in a variety of electronic formats. Some content that appears in print may not be available in electronic formats. For more information about Wiley products, visit our web site at www.wiley.com.
Library of Congress Cataloging-in-Publication Data:
Introduction to strategies for organic synthesis / Laurie S. Starkey.
p. cm.
ISBN 978-0-470-48409-8 (pbk.)
1. Organic compounds–Synthesis. I. Starkey, Laurie Shaffer, 1969–
QD262.I58 2012
547'.2–dc23
2011037217
ISBN 9780470484098
I really could have used this book when I started graduate school! I became fascinated with organic synthesis ever since running my first Grignard reaction as an undergraduate student at the University of Connecticut. As I watched the magnesium metal disappear into the solvent in my round-bottom flask, I was intrigued by the thought of making new molecules. Although my interest continued in graduate school at UCLA, I quickly found myself being thrown into the proverbial deep end when I took my first graduate course in organic synthesis. I had never taken a synthesis course at UConn, and my year of organic chemistry seemed like a foggy memory. I scoured every textbook I could find in an effort to stay afloat, but it was a struggle to work through the advanced material I found there. I appreciated the mentorship and patience of my research advisor, and I successfullly earned my Ph.D. in organic chemistry. Although I was able to make progress on my graduate research projects, I did not fully grasp the strategies of organic synthesis until I had to teach the course myself as a faculty member. As I embarked on my teaching career at Cal Poly Pomona, I was eager to share my passion for organic synthesis, but I found that most of my students were experiencing the same difficulties that I had encountered. The quantum leap from sophomore-level organic chemistry to senior-level organic synthesis is nearly insurmountable for some students. I did my best to bridge this gap, but it was a challenge since all of the available textbooks were written at the graduate level (or beyond!). After many years of teaching the organic synthesis course, I gradually developed a strategy that seemed to foster student success. My approach involves a significant amount of review of the sophomore-level material (functional group transformations, reagents, and reaction mechanisms) before changing the perspective and attempting to plan a synthesis (functional group analysis and making strategic disconnections: the retrosynthesis of a target molecule). Simply put, taking a year of organic chemistry does not make you an organic chemist, so this review is an essential element for most students. After enough experience, envisioning a reaction in both the forward and reverse directions is a routine exercise, but it cannot be assumed to be a trivial matter from the beginning. Such an assumption is made when little to no distinction is made between undergraduate-level and graduate-level organic synthesis courses, and it can result in a frustrating experience for the student. This book is designed as an intermediate-level introduction to the tools and skills needed to study organic synthesis. It contains worked-through examples and detailed solutions to the end-of-chapter problems, so it is ideal for any student who is interested in pursuing research in the field of organic chemistry, including beginning graduate students. With its thorough review of the reactions of organic chemistry and its study guide approach, this book can also build confidence as it deepens students’ knowledge and prepares them for advanced coursework.
To the students studying organic chemistry, I offer the same advice that I give to my kids (a quote from Mahatma Gandhi): “Live as if you were to die tomorrow. Learn as if you were to live forever.”
ACKNOWLEDGMENTS
I want to thank my students for making this the greatest job in the world; my graduate advisor, Mike Jung, for providing a supportive environment and for believing in a struggling student; and my mentor, Phil Beauchamp, who is both a passionate teacher and an organic synthesis junkie. The preparation of the manuscript would have been impossible without my tireless reviewers, including Phil Beauchamp, Joe Casalnuovo, Chris Nichols, Phil Lukeman, Richard Johnson, and especially Michael O’Donnell, an avid student of chemistry who somehow found my book before it was even finished! Most of all I am grateful to my wolfpack, Mike, Ellie, and Andy, because the most important things in life aren’t things.
Laurie S. Starkey
Cal Poly Pomona
CHAPTER 1.1
RETROSYNTHETIC ANALYSIS
Every organic synthesis problem actually begins at the end of the story, a target molecule. The goal is to design a reasonable synthesis that affords the target molecule as the major product. In the interest of saving both time and money, an ideal synthesis will employ readily available starting materials and will be as efficient as possible. The planning of a synthesis involves imagining the possible reactions that could give the desired target molecule product; this process is called doing a retrosynthesis or performing a retrosynthetic analysis of a target molecule. A special arrow is used to denote a retrosynthetic step. The ⇒ arrow leading away from the target molecule represents the question “What starting materials could I use to make this product?” and points to an answer to that question. The analysis begins by identifying a functional group (FG) present on the target molecule and recalling the various reactions that are known to give products containing that functional group (or pattern of FGs). The process is continued by analyzing the functional groups in the proposed starting material and doing another retrosynthetic step, continuing to work backwards toward simple, commercially available starting materials. Once the retrosynthetic analysis is complete, then the forward multistep synthesis can be evaluated, beginning with the proposed starting materials and treating them with the necessary reagents to eventually transform them into the desired target molecule.
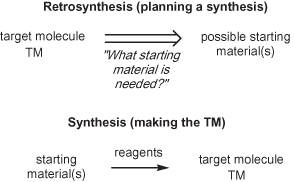
Retrosynthesis and Synthesis of a Target Molecule
A retrosynthesis involves working backwards from the given target molecule (work done in our minds and on paper), while the synthesis is the forward path leading to the target molecule (experimental work done in the lab). Performing a retrosynthetic analysis is challenging since it not only requires knowledge of an enormous set of known organic reactions, but also the ability to imagine the experimental conditions necessary to produce a desired product. This challenge becomes more manageable by developing a systematic approach to synthesis problems.*
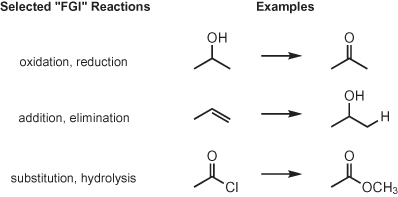
When evaluating a given target molecule, it is important to consider how the functional groups present in the target molecule can be formed. There are two possibilities for creating a given functional group: by conversion from a different functional group (called a functional group interconversion or FGI), or as a result of a bond-forming reaction (requiring a retrosynthetic “disconnection”). In order to synthesize a target molecule (or transform a given starting material into a desired product), a combination of FGIs and carbon–carbon bond-forming reactions will typically be required. While the key to the “synthesis” of complex organic molecules is the formation of new carbon–carbon bonds, the synthetic chemist must also be fully capable of swapping one functional group for another.
RETROSYNTHESIS BY FUNCTIONAL GROUP INTERCONVERSION (FGI)
Each functional group has a characteristic reactivity; for example, it might be electron-rich, electron-deficient, acidic, or basic. In order to synthesize organic compounds, we must construct the desired carbon framework while locating the required functional groups in the appropriate positions. This necessitates that the chemist is familiar not only with the reactivities of each functional group, but also the possible interconversions between functional groups. Such functional group interconversions enable the chemist to move along a synthetic pathway toward a desired target.
Examples of FGI
Let’s consider a carboxylic acid target molecule (RCO2H). There are many ways to generate a carboxylic acid functional group, so there are many possible syntheses to consider (often, there may be more than one good solution to a given synthesis problem!). One reaction that gives a carboxylic acid product is the hydrolysis of a carboxylic acid derivative, such as a nitrile. Therefore, a possible retrosynthesis of a carboxylic acid target molecule (What starting materials are needed?) is to consider a functional group interconversion and imagine a nitrile starting material. In other words, if we had a nitrile in our hands, we could convert it to a carboxylic acid, leading to a synthesis of the target molecule.
Retrosynthesis of a Target Molecule via FGI
Synthesis of the Target Molecule
Choice of Reagents
There is almost always more than one reagent that can be used to achieve any given transformation. In fact, a quick look at a book such as Comprehensive Organic Transformations by Richard Larock* reveals that there may be dozens of possibilities. Why have so many methods been developed over the years for organic reactions? Because not every molecule—or every chemist—has the same needs. The most obvious reason any “one size fits all” approach fails is that complex synthetic targets contain a wide variety of functional groups. The molecule as a whole must tolerate the reaction conditions used, and side reactions with other functional groups must be kept to a minimum. For example, chromic acid oxidation (Na2Cr2O7, H2SO4) of a 2° alcohol to give a ketone would not be useful if the starting material contains any functional groups that are sensitive to acidic conditions. In such a case, the Swern oxidation might be preferred (DMSO, ClCOCOCl, Et3N). New reagents, catalysts, and methods are continuously being developed, with goals of having better selectivity, better tolerance for certain functional groups, being “greener” with less waste or lower toxicity, requiring fewer steps, being more efficient and/or less expensive, and so on.
The focus of this book is on the strategies of organic synthesis; it is not intended to be comprehensive in the treatment of modern reagents.* Instead, reagents used are those that are typically found in undergraduate organic chemistry textbooks. Hopefully, these reagents will be familiar to the reader, although they would not necessarily be the ones selected when the synthesis moves from paper to the laboratory. Furthermore, experimental details† have largely been omitted from this book. For example, osmium tetroxide oxidation of an alkene is given simply as “OsO4.” In reality, this expensive and toxic reagent is used in catalytic amounts in conjunction with some other oxidizing agent (e.g., NMO), so the precise reagents and experimental reaction conditions are much more complex than what is presented herein.
RETROSYNTHESIS BY MAKING A DISCONNECTION
Rather than being created via a functional group interconversion, a functional group (or pattern of functional groups) may be created as a result of a reaction that also forms a carbon–carbon sigma bond. In that case, the retrosynthesis involves the disconnection of that bond. In a typical carbon–carbon bond-forming reaction, one of the starting material carbons must have been a nucleophile (Nu:, electron-rich), and the other must have been an electrophile (E+, electron-deficient). While this is certainly not the only way to make a carbon–carbon bond (e.g., organometallic coupling reactions), the pairing of appropriate nucleophiles and electrophiles serves as an important foundation to the logic of organic synthesis, and such strategies will solve a wide variety of synthetic problems. Therefore, the disconnection of the carbon–carbon bond is made heterolytically to give an anion (nucleophile) and a cation (electrophile). These imaginary fragments, called “synthons,” are then converted into reasonable starting materials. By being familiar with common nucleophiles and electrophiles, we can make logical disconnections. The example below shows the logical disconnection of an ether target molecule, affording recognizable alkyl halide E+ and alkoxide Nu: starting materials.
A Logical Disconnection of a Target Molecule
Disconnecting that same carbon–oxygen bond in the other direction (with both electrons going to the carbon) would be an illogical disconnection, since it leads to an electrophilic oxygen synthon for which there is no reasonable equivalent reagent.
An Illogical Disconnection of a Target Molecule
Let’s consider once again a carboxylic acid target molecule. We’ve seen that a carboxylic acid can be prepared by an FGI if the carbon chain is already in place, but it is also possible to create new carbon–carbon bonds in a carboxylic acid synthesis. For example, the reaction of a Grignard reagent with carbon dioxide generates a carboxylic acid functional group, so this presents a possible disconnection for the target molecule’s retrosynthesis. The logical disconnection is the one that moves the electrons away from the carbonyl, giving reasonable synthons and recognizable starting materials (RMgBr Nu: and CO2 E+).
Retrosynthesis via Disconnection of a Target Molecule
Synthesis of the Target Molecule
What Makes a Good Synthesis?
The fact that multiple retrosynthetic strategies usually exist means that there will often be more than one possible synthesis of a desired target molecule. How can we determine which synthesis is best? This depends on many factors, but there are some general rules that can help us devise a good plan to synthesize the simple target molecules found in this book.
1.
Start with reasonable starting materials and reagents. A good synthesis begins with commercially available starting materials. Most of these starting materials will have a small number of functional groups (just one or two), although some complex natural products are readily available and inexpensive (e.g., sugars and amino acids). A quick check in any chemical supplier catalog can confirm whether a starting material is ordinary (i.e., available and inexpensive) or exotic (i.e., expensive or not listed).
2.
Propose a reaction with a reasonable reaction mechanism. Look for familiar nucleophiles and electrophiles to undergo predictable reactions. A poor choice for a bond disconnection can lead to impossible synthons (and impossible reagents). However, we will learn that certain seemingly impossible synthons are, in fact, possible with the use of synthetic equivalents.
3.
Strive for disconnections that lead to the greatest simplification. It is bad practice to put together a 10-carbon target molecule one carbon at a time (an example of a linear synthesis). Remember, the synthetic schemes drawn on paper represent reactions that will be performed in the lab. While this book will not be focusing on experimental details, we should recognize that the more steps in a reaction sequence, the lower the overall yield of product will be. Starting with a nine-carbon starting material, which is nearly as big and possibly as complicated as a 10-carbon target molecule, also would not be a good synthesis. The most efficient synthesis would be one that links together two five-carbon structures, or perhaps one that combines a four-carbon with a six-carbon compound (described as a convergent synthesis). The more nearly equal the resulting pieces, the better the bond disconnection. One useful strategy is to look for branch points in a target molecule for good places to make a disconnection. In the example below, the starting materials resulting from disconnection “a” are not only more simple molecules, but also the butanal starting material (butyraldehyde) is one-tenth the price of the aldehyde in disconnection “b” (2-methylbutyraldehyde).
Good Disconnections Lead to Simple, Inexpensive Starting Materials
Notes
* For the classic textbook on such an approach, see Stuart Warren and Paul Wyatt, Organic Synthesis: The Disconnection Approach, 2nd ed. (Wiley, 2009).
* Richard C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 2nd ed. (Wiley-VCH, 1999).
* Tse-Lok Ho, Fieser and Fieser’s Reagents for Organic Synthesis Volumes 1–26, and Collective Index for Volumes 1–22, Set, 1st ed. (Wiley, 2011); Leo A. Paquette et al., Encyclopedia of Reagents for Organic Synthesis, 14 Volume Set, 2nd ed. (Wiley, 2009); George Zweifel and Michael Nantz, Modern Organic Synthesis, 1st ed. (W. H. Freeman, 2006).
† A.I. Vogel et al., Vogel’s Textbook of Practical Organic Chemistry, 5th ed. (Prentice Hall, 1996).


 C (alkyne), OH (alcohol or carboxylic acid), and C
C (alkyne), OH (alcohol or carboxylic acid), and C O (aldehyde, ketone, and many others). The process of planning a synthesis, called a retrosynthesis, is one of the most critical tools within the “toolbox” needed to solve synthesis problems. The method of retrosynthetic analysis is introduced in this chapter and is used throughout the book. This first chapter will also review the use of protective groups (PGs) in organic synthesis. The second chapter provides additional useful tools needed by the beginning synthesis student by reviewing common nucleophiles and electrophiles, as well as some general reagents for oxidation and reduction reactions.
O (aldehyde, ketone, and many others). The process of planning a synthesis, called a retrosynthesis, is one of the most critical tools within the “toolbox” needed to solve synthesis problems. The method of retrosynthetic analysis is introduced in this chapter and is used throughout the book. This first chapter will also review the use of protective groups (PGs) in organic synthesis. The second chapter provides additional useful tools needed by the beginning synthesis student by reviewing common nucleophiles and electrophiles, as well as some general reagents for oxidation and reduction reactions.