
Contents
Preface
General References
1 The Disconnection Approach
A Synthesis of Multistriatin
References
2 Basic Principles: Synthons and Reagents: Synthesis of Aromatic Compounds
A Problem from the Textbook
References
3 Strategy I: The Order of Events
References
4 One-Group C–X Disconnections
References
5 Strategy II: Chemoselectivity
Using Disconnections to Solve Structural and Mechanistic Problems
References
6 Two-Group C–X Disconnections
Counting Relationships between Functional Groups
Synthesis of a Heterocycle
References
7 Strategy III: Reversal of Polarity, Cyclisations, Summary of Strategy
Regioselective Attack on Epoxides
References
8 Amine Synthesis
An Example of a Triamine
Strategic Bond Disconnection
A New Generation Pfizer anti-HIV Drug Maraviroc
References
9 Strategy IV: Protecting Groups
Synthesis without Protection
Protection
An HIV-Protease Inhibitor as an anti-AIDS Drug
Example: Synthesis of Statins (Cholesterol-Lowering Drugs)
References
10 One-Group C–C Disconnections I: Alcohols
An Example of Simple Alkylation
References
11 General Strategy A: Choosing a Disconnection
Summary of Guidelines for Good Disconnections
The Synthesis of an Unusual Amino Acid
Strategy in the Synthesis of Sildenafil (Viagra®)
References
12 Strategy V: Stereoselectivity A
References
13 One-Group C–C Disconnections II: Carbonyl Compounds
References
14 Strategy VI: Regioselectivity
Other Examples of Regioselectivity
A Heterocyclic Example
References
15 Alkene Synthesis
A Pharmaceutical Example
The Importance of Experimental Work
References
16 Strategy VII: Use of Acetylenes (Alkynes)
Examples from the Textbook Chapter
Synthesis of a Cyclic Ketone by Hydration of an Acetylene
An Interesting Mechanism and a Useful Separation
Electrophilic Acetylenes
Alkynes in Synthesis
References
17 Two-Group C–C Disconnections I: Diels-Alder Reactions
References
18 Strategy VIII: Introduction to Carbonyl Condensations
References
19 Two-Group C–C Disconnections II: 1,3-Difunctionalised Compounds
A Synthesis of the Enzyme Inhibitor Elasnin
References
20 Strategy IX: Control in Carbonyl Condensations
Three Examples
References
21 Two-Group C–C Disconnections III: 1,5-Difunctionalised Compounds Conjugate (Michael) Addition and Robinson Annelation
References
22 Strategy X: Aliphatic Nitro Compounds in Synthesis
The Synthesis of an ACE Inhibitor
References
23 Two-Group Disconnections IV: 1,2-Difunctionalised Compounds
Acyl Anion Equivalents
Some Problems
α-Functionalisation of Carbonyl Compounds
References
24 Strategy XI: Radical Reactions in Synthesis
The Mechanism of Allylic Bromination with NBS
Application of NBS in Synthesis
Carbon–Carbon Bond-Forming Reactions
A Pharmaceutical Example
References
25 Two-Group Disconnections V: 1,4-Difunctionalised Compounds
Buying the 1,4-diCO Relationship
Troubles and Triumphs with Homoenolates
A General Synthesis of Partly Protected Succinic Acids
A Remarkable Reaction from the Textbook
References
26 Strategy XII: Reconnection
Synthesis of 1,2-and 1,4-diCO Compounds by Oxidative C=C Cleavage
Oxidative Cleavage of Aldol Products
Cleavage of Aldol Products by Retro-Aldol Reaction
References
27 Two-Group C–C Disconnections VI: 1,6-diCarbonyl Compounds
Problems from the Textbook
The Synthesis of Acorenone B
The Synthesis of a Symmetrical Keto-di-Acid
Oxidative Cleavage by the Baeyer-Villiger Rearrangement
References
28 General Strategy B: Strategy of Carbonyl Disconnections
The Synthesis of Long Chain Fatty Acids
The Synthesis of a Furan
The Synthesis of a Modern Drug Candidate
References
29 Strategy XIII: Introduction to Ring Synthesis: Saturated Heterocycles
Cyclisation Reactions
A Bicyclic Amine
References
30 Three-Membered Rings
Cyclisation and Carbene Strategies Compared
Cyclopropanes from Electrophilic Alkenes
The Synthesis of Halicholactone
References
31 Strategy XIV: Rearrangements in Synthesis
Diazoalkanes
The Pinacol Rearrangement
The Favorskii Rearrangement
References
32 Four-Membered Rings: Photochemistry in Synthesis
An Example from Chapter 31
Development of Material from the Textbook
Photochemical Cycloadditions
Four-Membered Rings by Ionic Reactions
References
33 Strategy XV: The Use of Ketenes in Synthesis
Do Ketenes Exist?
The Synthesis of α-Lactones
Ketenes as Intermediates
[2 + 2] Thermal Cycloadditions of Ketenes
References
34 Five-Membered Rings
An Intermediate in the Synthesis of Coriolin
Asymmetric Synthesis from Terpenes
Cyclisation of Alkyl Lithiums onto Alkenes
References
35 Strategy XVI: Pericyclic Reactions in Synthesis: Special Methods for Five-Membered Rings
Electrocyclic Reactions
Sigmatropic Rearrangements
References
36 Six-Membered Rings
A Synthesis from the Textbook Chapter
The Diels-Alder Route
The Birch Reduction Route
References
37 General Strategy C: Strategy of Ring Synthesis
Development of Some Chemistry from the Textbook
References
38 Strategy XVII: Stereoselectivity B
The Prelog-Djerassi Lactone
A Pharmaceutical Example
The Synthesis of a Cage Molecule
Conformational Control
References
39 Aromatic Heterocycles
The Mechanism of the Stetter Synthesis of 1,4-diCarbonyl Compounds
The Synthesis of Five-Membered Heterocycles
Mechanisms in Heterocyclic Chemistry
Pyrazole, Imidazole and Quinoline
References
40 General Strategy D: Advanced Strategy
The Synthesis of Methoxatin
The Key Reaction Strategy: Diels-Alder Reactions
References
Index

This edition first published 2009
© 2009 John Wiley & Sons Ltd
Registered office
John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, United Kingdom
For details of our global editorial offices, for customer services and for information about how to apply for permission to reuse the copyright material in this book please see our website at www.wiley.com.
The right of the author to be identified as the author of this work has been asserted in accordance with the Copyright, Designs and Patents Act 1988.
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs and Patents Act 1988, without the prior permission of the publisher.
Wiley also publishes its books in a variety of electronic formats. Some content that appears in print may not be available in electronic books.
Designations used by companies to distinguish their products are often claimed as trademarks. All brand names and product names used in this book are trade names, service marks, trademarks or registered trademarks of their respective owners. The publisher is not associated with any product or vendor mentioned in this book. This publication is designed to provide accurate and authoritative information in regard to the subject matter covered. It is sold on the understanding that the publisher is not engaged in rendering professional services. If professional advice or other expert assistance is required, the services of a competent professional should be sought.
The publisher and the author make no representations or warranties with respect to the accuracy or completeness of the contents of this work and specifically disclaim all warranties, including without limitation any implied warranties of fitness for a particular purpose. This work is sold with the understanding that the publisher is not engaged in rendering professional services. The advice and strategies contained herein may not be suitable for every situation. In view of ongoing research, equipment modifications, changes in governmental regulations, and the constant flow of information relating to the use of experimental reagents, equipment, and devices, the reader is urged to review and evaluate the information provided in the package insert or instructions for each chemical, piece of equipment, reagent, or device for, among other things, any changes in the instructions or indication of usage and for added warnings and precautions. The fact that an organization or Website is referred to in this work as a citation and/or a potential source of further information does not mean that the author or the publisher endorses the information the organization or Website may provide or recommendations it may make. Further, readers should be aware that Internet Websites listed in this work may have changed or disappeared between when this work was written and when it is read. No warranty may be created or extended by any promotional statements for this work. Neither the publisher nor the author shall be liable for any damages arising herefrom.
Library of Congress Cataloging-in-Publication Data
Warren, Stuart.
Workbook for organic synthesis: the disconnection approach/Stuart Warren and Paul Wyatt. – 2nd ed.
p. cm.
Includes bibliographical references and index.
ISBN 978-0-470-71227-6 – ISBN 978-0-470-71226-9
1. Organic compounds – Synthesis – Textbooks. I. Wyatt, Paul. II. Title.
QD262.W93 2009
547'.2 – dc22
2009030810
A catalogue record for this book is available from the British Library.
ISBN 978-0-470-7-12276 (h/b) 978-0-470-7-12269 (p/b)
Preface
In the 26 years since Wiley published Organic Synthesis: The Disconnection Approach and the accompanying Workbook, this approach to the learning of synthesis has become widespread while the books themselves are now dated in content and appearance. In 2008, Wiley published the second edition of Organic Synthesis: The Disconnection Approach by Stuart Warren and Paul Wyatt for which this is the accompanying Workbook.
This workbook contains further examples, problems (and answers) to help you understand the material in each chapter of the textbook. The structure of this second edition of the workbook is the same as that of the textbook. The 40 chapters have the same titles as before but all chapters have undergone a thorough revision with some new material. The emphasis is on helpful examples and problems rather than novelty. Many of the problems are drawn from the courses we have given in industry on ‘The Disconnection Approach’ where they have stimulated discussion leading to deeper understanding. It makes sense for you to have the relevant chapter of the textbook available while you are working on the problems. We have usually devised new problems but some of the problems in the first edition seemed to do such a good job that we have kept them. Usually, the answers are presented in a different and, we hope, more helpful style.
It is not possible to learn how to design organic syntheses just from lectures or from reading a textbook. Only by tackling problems and checking your answers against published material can you develop this skill. We should warn you that there is no single ‘right answer’ to a synthesis problem. Successful published syntheses give some answers that work, but you may well be able to design others that have a good chance of success. The style of this second edition is to give more discussion of alternative routes.
Stuart Warren and Paul Wyatt
2009
General References
Full details of important books referred to by abbreviated titles in the chapters to avoid repetition.
Clayden Organic Chemistry: J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic Chemistry, Oxford University Press, Oxford, 2000.
Disconnection Textbook: S. Warren and P. Wyatt, Organic Synthesis: The Disconnection Approach, Second Edition, Wiley, Chichester, 2008.
Drug Synthesis: D. Lednicer and L. A. Mitscher, The Organic Chemistry of Drug Synthesis, Wiley, New York, seven volumes, from 1977.
Fieser, Reagents: L. Fieser and M. Fieser, Reagents for Organic Synthesis, Wiley, New York, 20 volumes, 1967–2000, later volumes by T.-L. Ho.
Fleming, Orbitals: Ian Fleming, Frontier Orbitals and Organic Chemical Reactions, Wiley, London, 1976.
Vogel: B. S. Furniss, A. J. Hannaford, P. W. G. Smith, and A. R. Tatchell, Vogel’s Textbook of Practical Organic Chemistry, Fifth Edition, Longman, Harlow, 1989.
1
The Disconnection Approach
We start with a few simple problems to set you at ease with disconnections. Problem 1.1: Here is a two-step synthesis of the benzofuran 3. Draw out the retrosynthetic analysis for the synthesis of 2 from 1 showing the disconnections and the synthons.
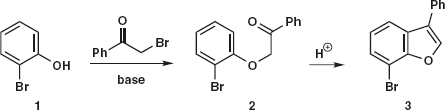
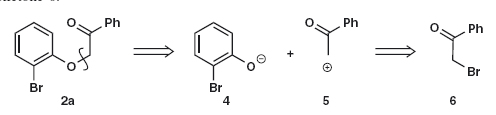
Answer 1.1: As this is a simple SN2 reaction, the disconnection is of the C–O bond 2a and the synthons are nucleophilic phenolate anion 4, which happens to be an intermediate in the reaction, and the cation 5, which happens not be an intermediate in the reaction but is represented by the α-bromoketone 6.
Problem 1.2: Draw the mechanism of the cyclisation of 2 to 3. This is an unusual reaction and it helps to know what is going on before we analyse the synthesis. Answer 1.2: The first step is an acid-catalysed cyclisation of the aromatic ring onto the protonated ketone 7. Loss of a proton 8 completes the electrophilic aromatic substitution giving the alcohol 9.
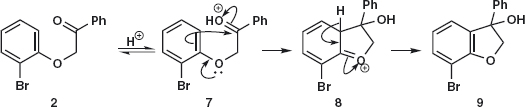
Now protonation of the alcohol leads to loss of water 10 to give a stabilised cation that loses aproton 11 to give the new aromatic system 3. Problem 1.3: Now you should be in a position to draw the disconnections for this step.

Answer 1.3: We hope you might have drawn the intermediate alcohol 9. Changing 3 into 9 is not a disconnection but a Functional Group Interconversion (FGI) – changing one functional group into another. Now we can draw the disconnection revealing the synthons 12 represented in real life by 2.

A Synthesis of Multistriatin
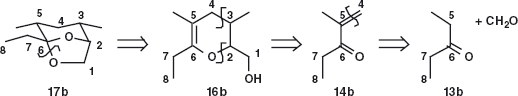
In the textbook we gave one synthesis of multistriatin 17 and here is a shorter but inferior synthesis as the yields are lower and there is little control over stereochemistry.1 Problem 1.4: Which atoms in the final product 17 come from which starting material and which bonds are made in the synthesis? Hint: Arbitrarily number the atoms in multistriatin and try to trace each atom back through the intermediates. Do not be concerned over mechanistic details, especially of the step at 290°C.
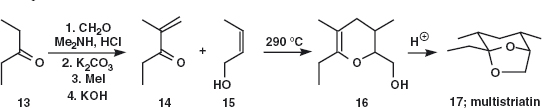
Answer 1.4: However you numbered multistriatin, the ethyl group (7 and 8 in 17a) finds the same atoms in the last intermediate 16a and the rest falls into place. It then follows which atoms come from 14 and which from 15. Finally, you might have said that C-4 in our diagrams comes from formaldehyde.

So the disconnections also fall into place. Just one C–O bond was disconnected at first 17b then one C–O and one C–C 16b and finally the alkene was disconnected 14b in what you may recognise as an aldol reaction with formaldehyde. If you practise analysing published syntheses like this, you will increase your understanding of good bonds to disconnect.
References
1. W. E. Gore, G. T. Pearce and R. M. Silverstein, J. Org. Chem., 1975, 40, 1705.
2
Basic Principles: Synthons and Reagents: Synthesis of Aromatic Compounds
This chapter concerns the synthesis of aromatic compounds by electrophilic and nucleophilic aromatic substitution. All the disconnections will therefore be of bonds joining the aromatic rings to the sidechains. We hope you will be thinking mechanistically, particularly when choosing which compounds can undergo nucleophilic aromatic substitution and the orientation of electrophilic aromatic substitution. Any textbook of organic chemistry1 will give you the help you need.
Problem 2.1: Compound 1 was needed2 for an exploration of the industrial uses of HF. Suggest how it might be made. Hint: consider which of the three substituents you would rather not add to the ring.

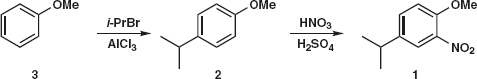
Answer 2.1: We can add the nitro group by nitration and the isopropyl group by Friedel-Crafts alkylation (as it is a secondary alkyl group) but we would rather not add the OMe group as there is no good reagent for MeO+. So we disconnect first the most deactivating group (nitro) 1a and then the isopropyl group 2.
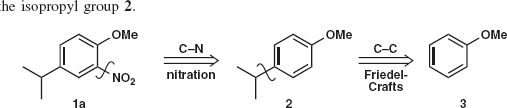
Before writing out the synthesis, we should check that the orientation of the substitution will be what we want. The OMe group is ortho, para-directing so alkylation will go mainly para because of steric hindrance. Now we have a competition as isopropyl is also ortho, para-directing but, since OMe has a lone pair of electrons conjugated with the benzene ring, it will dominate so everything is fine. We therefore suggest:
Did you consider the alternative strategy? That is, disconnect the isopropyl group first 1b to give a new intermediate 4 and disconnect the nitro group second. The starting material, anisole 3, is the same in both routes.
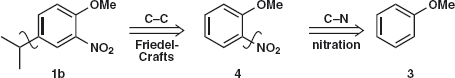
Again we should check the orientation. Nitration of anisole will give a mixture of ortho 4 and para 5 products so much depends on the ratio and whether they can easily be separated. The Friedel-Crafts reaction will go ortho or para to the OMe group and meta to the nitro group so that is all right. However the deactivating nitro group might make the reaction difficult.
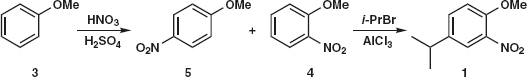
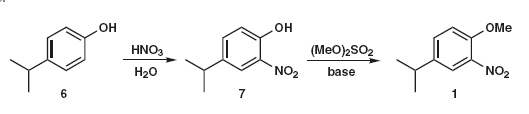
So what did the chemists prefer? One published synthesis2 used HF as a catalyst to alkylate ortho-nitro-anisole 4 with isopropanol. The yield was a respectable 84%. This made sense as they had a supply of 4. If anisole is nitrated with the usual HNO3 /H2 SO4 , a 31:67 ratio of ortho:para products is obtained. If the nitrating agent is an alkyl nitrite in MeCN, the ratio improves to 75:25. The best route nowadays is probably the nitration of available para-isopropyl phenol 6, probably quantitative, and methylation of the product 7 with, say, dimethyl sulfate.
Problem 2.2: These compounds 8 and 9 each have two benzene rings linked by a heteroatom and both are used to make anti-inflammatory drugs. An obvious strategy is to disconnect one C–X bond in each case and combine the two compounds by nucleophilic aromatic substitution. Suggest a synthesis for each compound.
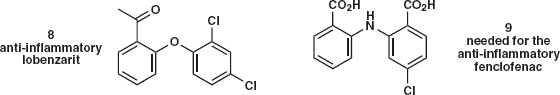
Answer 2.2: The two disconnections 8a and 8b illustrate the types of molecules needed for the first problem. In each case X is a leaving group such as a halogen and the phenols 11 and 12 would be used as their anions.
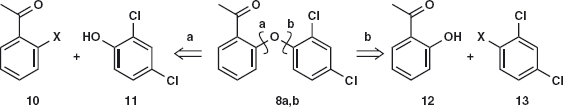
To be successful, nucleophilic aromatic substitution needs an electron-withdrawing group ortho or para to the leaving group. A chloride, as in 13 is not adequate but the ketone in 10 is perfectly placed. The reported synthesis3 uses 10;X = Cl with 11 and Cu/NaOH as catalyst. We might nowadays prefer available 10;X = F with the anion of the phenol.
The other compound 9 is easier in one way as both disconnections 9a and 9b are feasible. Each ring 14 and 15 has an electron-withdrawing CO2 H group in the right position (ortho to the leaving group X). Compound 17 has another leaving group (Cl) that is para to the CO2 H group so it could react. On the other hand, compound 15 could react with itself and polymerise as it has the nucleophilic amine and the activated chloride in the same molecule.
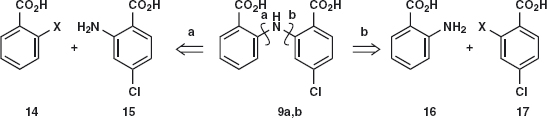
The reported synthesis4 uses 16 and 17; X = Cl relying on the CO2 H group to provide regioselectivity at the more electrophilic ortho position. It is possible5 that the fluoro-compound 17; X = F would be a better way.
Problem 2.3: Chagas disease causes some 50,000 deaths annually in South America. Drugs based on the structure 18 are urgently needed. You are not expected to understand the chemistry used to make the strange heterocyclic ring but you might appreciate that it could come from an ortho-nitro aniline such as 19 or an activated halide such as 20. Suggest syntheses for these starting materials.
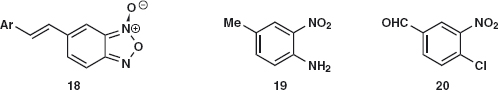
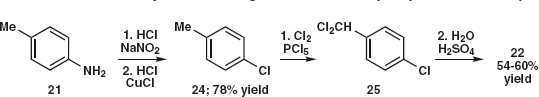
Answer 2.3: In both cases, the initial disconnection of the nitro group 19a and 20a is very appealing. The starting materials 21 and 22 should be easily made and nitration will go ortho to NH2 rather than Me in 21 and ortho to Cl and meta to the deactivating aldehyde in 22.
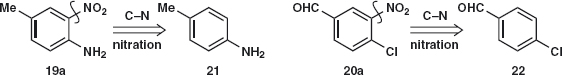
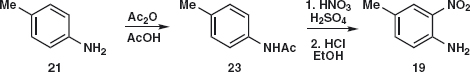
The synthesis of 19 is straightforward6 as the amine 21 is available from the nitration and reduction of toluene. Amide 23 formation reduces the reactivity of the amine so that mono-nitration and hydrolysis give 19. Nitration of 23 gives 19.
The aldehyde 22 is more difficult as we should need to chlorinate benzaldehyde in the para position to get 22. One solution is to oxidise para chloro-toluene 24, available7 from 21 via the diazonium salt with, for example, chlorine to give 25 that can be hydrolysed8 to the aldehyde 22.
A Problem from the Textbook
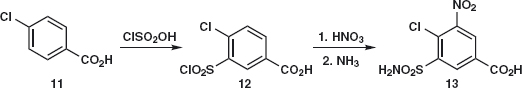
When discussing the synthesis of saccharine in chapter 2 of the textbook, we said; ‘In practice chloro-sulfonic acid is used as this gives the sulfonyl chloride directly. You may be surprised at this, thinking that Cl might be the best leaving group. But there is no Lewis acid here. Instead the very strong chloro-sulfonic acid protonates itself to provide a molecule of water as leaving group.’ The reaction gives a mixture of the ortho-27 and para-28 products. Problem 2.4: With those hints, draw a mechanism of the chlorosufonation.
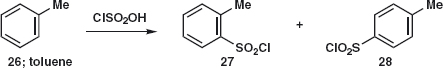
Answer 2.4: ‘Strong’ means a strong acid here so chloro-sulfonic acid 29 protonates itself to give a cation that loses water 30 to give the reactive cation 31. This is attacked by toluene in the ortho-and para-positions to give e.g. 32 that loses a proton to give 28.
References
1. Clayden, Organic Chemistry, chapters 22 and 23.
2. W. S. Calcott, J. M. Tinker and V. Weinmayr, J. Am. Chem. Soc., 1939, 61, 1010.
3. Drug Synthesis, vol 4, p. 42.
4. Drug Synthesis, vol 3, p. 38.
5. S. M. Kelly and H. Schad, Helv. Chim. Acta, 1985, 68, 1444.
6. W. Porcal, A. Merlino, M. Boiani, A. Gerpe, M. Gonz´alez and H. Cercetto, Org. Process. Res. Dev., 2008, 12, 156.
7. Vogel, p. 931.
8. W. L. McEwen, Org. Synth. Coll., 1943, 2, 133.
3
Strategy I: The Order of Events
You should refer to the Guidelines from the textbook when you solve the problems in this chapter.
Guideline 1: Consider the effects of each functional group on the others. Add first (that is disconnect last) the one that will increase reactivity in a helpful way.
Guideline 2: Changing one functional group into another may alter reactivity dramatically.
Guideline 3: Some substituents are difficult to add so it is best to start with them already present.
Guideline 4: Some disubstituted compounds are also readily available and they may contain a relationship (especially ortho) that is difficult to achieve by electrophilic substitution.
Guideline 5: Some groups can be added to the ring by nucleophilic substitution.
Guideline 6: If a series of reactions must be carried out, start with one that gives a single product unambiguously and not one that would give a mixture.
Remember that these guidelines may conflict or even contradict each other. THINK! Problem 3.1: Suggest syntheses of 1 and 2 needed as intermediates: 1 in the synthesis of some brominated acids1 and 2 to study the mechanism of enzymatic ester hydrolysis.2

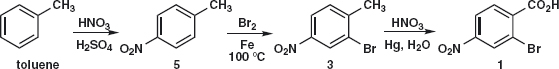
Answer 3.1: With two electron-withdrawing groups in 1, some FGI is needed to control the orientation and gain some reactivity. There are good ways to introduce Br and NO2 but no easy way to introduce CO2H. FGI of CO2H to Me with oxidation in mind would give an ortho, para-directing group where we need it 3. Now we might disconnect NO2 3a or Br 3b as there are good reagents for adding both. There might be some doubt as to where 4 would be nitrated as both Me and Br are ortho, para-directing, but there is no doubt where 5 will be brominated as Me is ortho, para-directing while NO2 is meta-directing.
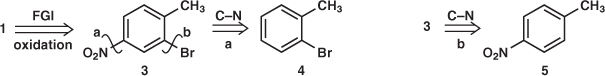
So the synthesis was nitration of toluene (actually 5 is available), separation of 5 from the ortho isomer, bromination of 5, and oxidation of 3 to give the target molecule.1
No doubt the CHO group could also be formed by oxidation of a CH3 group but it can be inserted next to a phenolic OH by the Reimer-Tiemann reaction.3 Now we can disconnect the t-Bu group with Friedel-Crafts alkylation in mind.
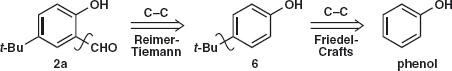
The large t-Bu group much prefers the para position and the Reimer-Tiemann reaction using chloroform as a source of dichlorocarbene (Textbook chapter 30) goes ortho to the conjugating OH group.2,4
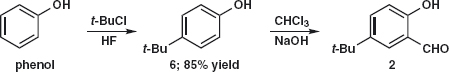
Example and Problem 3.2: Bumetamide 7 is a diuretic from Leo Pharmaceutical Products in Denmark. The synthesis5 was planned by a number of FGIs to give 8 and then a C–O disconnection to give 9 as a suitable starting material. Problem 3.2: Suggest why these FGIs were chosen as a preliminary to disconnection.
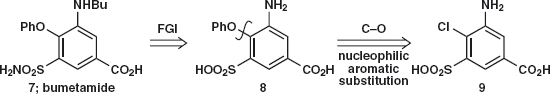
Answer 3.2: The PhO group must be added by nucleophilic aromatic substitution so electron-withdrawing groups are essential. We have two (SO2X and CO2H) in the right positions, ortho and para to Cl in 9, and could have a third if NH2 is replaced by NO2. Problem 3.3: Suggest a synthesis of 9.
Answer 3.3: Two of the substituents in 9 (SO2OH and Cl) can be added by electrophilic substitution and we have seen some ways to add the CO2H group. The most obvious thing to do is to replace NH2 by NO2 10 and disconnect both NO2 and SO2OH giving p-chlorobenzoic acid 11 as starting material. This compound is available but could be made by chlorination of toluene and oxidation of the methyl group.
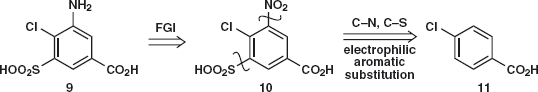
Now we need to decide in which order to add the two substituents. The orientation will be decided by the Cl group as it is ortho, para-directing. In the published synthesis5 chlorosulfonation is used followed by nitration and the sulfonamide 13 is formed before the nitro group is reduced to the amine.
With three groups to help nucleophilic substitution, phenoxide was added and catalytic hydrogenation of 14 to the amine 15 was followed by reductive amination (chapter 8) with PrCHO to give bumetamide 7.
References
1. K. Friedrich and H. Oster, Chem. Ber., 1961, 94, 834.
2. R. Breslow, M. F. Czarniecki, J. Emert and H. Hamaguchi, J. Am. Chem. Soc., 1980, 102, 762.
3. Vogel, pp. 992 and 997.
4. J. H. Simons, S. Archer and H. J. Passino, J. Am. Chem. Soc., 1938, 60, 2956.
5. P. W. Feit, H. Bruun and C. K. Nielsen, J. Med. Chem., 1970, 13, 1071; P. W. Feit, Ibid., 1971, 14,
4
One-Group C–X Disconnections
If you have also read chapter 6, you will realise that acid derivatives such as esters 1 or amides 3 are usually made by acylation so that the C–O or C–N bond that is disconnected is the one between the heteroatom and the carbonyl group. In this way we are really using two-group disconnections for these compounds. The synthesis might combine an alcohol or an amine with an acid chloride 2.

Problem 4.1: Suggest which C–X bond would be your first choice for disconnection in these two compounds, explaining your reasons. Draw your proposed starting materials.
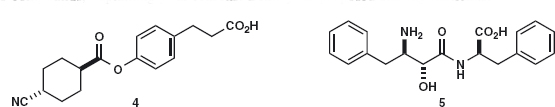
Answer 4.1: Though there are many C–X bonds in both molecules, the first disconnection should be of the ester 4a and of the amide 5a both because we know of good ways to make these functional groups and because the disconnections are in the middle of the molecules. You might have drawn 6 and 8 as acid chlorides or as acids, as we have done, deciding to work out the reagents later. Problem 4.2: What difficulties do you foresee in carrying out the reaction?
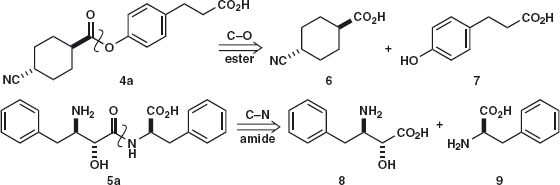
Answer 4.2: Both 6 and 7 have acid groups, so we shall have to activate the CO2H group in 6 and perhaps protect the CO2H group in 7. The situation for 8 + 9 is worse: not only does each compound have a CO2H group, but 8 also has two nucleophilic groups (OH and NH2). Again protection and activation will be needed. This second case is not as bad as it seems as 5 is a dipeptide and standard peptide coupling procedures can be used.1 Stereochemistry is not a problem as the bond-forming steps do not affect any chiral centre.
We shall concentrate mainly on ethers and sulfides where true one-group C–X disconnections will be needed though mechanistic arguments will still be necessary. Problem 4.3: Suggest a synthesis for the ethers 10 and 11.
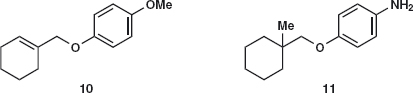
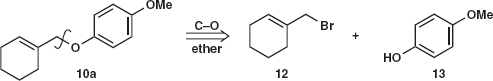
Answer 4.3: The first 10 is easy: we much prefer the disconnection on the alkyl side as the aromatic ring is not activated for nucleophilic substitution while the halide 12 is allylic and therefore electrophilic.
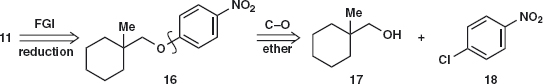
The second 11 requires more thought: The same disconnection 11a gives a primary halide 14 but it has a quaternary centre joined to it and there will be considerable steric hindrance to an SN2 reaction. In addition, the amine in 15 is more nucleophilic than the phenolic OH group. Is there an alternative?

The amine 11 could be made by reduction of a nitro group and now the alternative disconnection 16 corresponding to nucleophilic aromatic substitution becomes possible.2 There is no longer any ambiguity as there is only one nucleophilic group. In addition, the halide 14 would have to be made from the alcohol 17. Compounds derived from 11 are used in the treatment of diabetes.
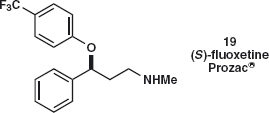
Problem 4.4: Suggest a synthesis of fluoxetine 19, better known as the antidepressant Prozac®.
Answer 4.4: We should rather disconnect the ether in the middle of the molecule than the amine at the end and here again either C–O disconnection 19a or 19b would serve. The electrophile 21 (X is a leaving group) is benzylic and reactive while the CF3 group activates the ring for SNAr by stabilising the intermediate anion in the same way as the nitro group.
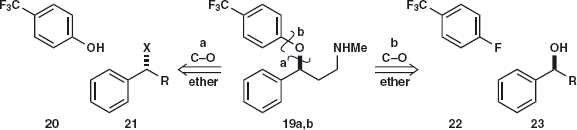
No doubt either synthesis will work but we could consider that the reaction at the chiral centre 19a might lead to some racemisation while reaction of 23 does not involve the chiral centre. The synthesis has been carried out with a single enantiomer of 23 using NaH as base in an amide solvent.3 The base gives the anion 24 so that oxygen becomes more nucleophilic than nitrogen.
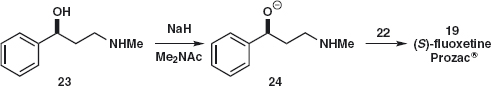
The question remains: how do we make the aminoalcohol 23? Using a one-group disconnection of the C–N bond, we can displace a leaving group X from 25 and a search of available starting materials reveals the chloroketone 26.
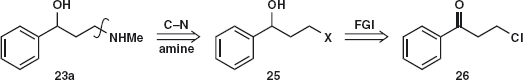
Reduction of the ketone and reaction of the chloride 27 with NaI in acetone (NaCl precipitates from acetone and drives the equilibrium to the right) gave the corresponding iodide 28. Reaction of 28 with an excess of MeNH2 as its available aqueous solution gave 23 in quantitative yield.3
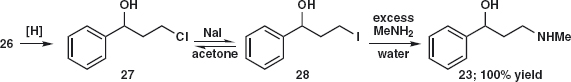
An alternative is to add the second aromatic ring by a Mitsunobu reaction and displace chloride afterwards with aqueous MeNH2. If a single enantiomer, e.g. (R)-(+)-27, is used, the inverted product (S )-(–)-29 is formed stereospecifically by the Mitsunobu reaction.4
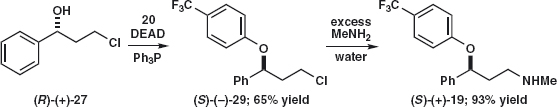
A related route starts with the epoxidation of cinnamyl alcohol 30 and regioselective reduction of the epoxide 31 by Red-Al, NaH2Al(OCH2CH2OMe)2 to give 32 because the aluminium complexes to the primary alcohol and delivers hydride to the nearer end of the epoxide. Mesylation and displacement with aqueous MeNH2 complete the synthesis.5
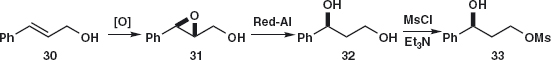
Problem 4.5: Suggest a synthesis of febantel 34 used as an anthelmintic to combat tapeworms and the like.

Answer 4.5: If we do the obvious amide disconnection first 34a, we have a serious problem of chemoselectivity as we shall have to acylate one of two very similar amines 35.But if we change the other amine into a nitro group 36, the problem disappears and also suggests how we might make the sulfide.
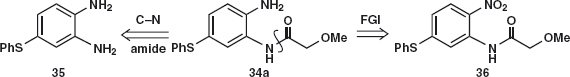
Amide disconnection 36a reveals a simple nitro-amine with the PhS group in just the right position for C–S disconnection 37 with the nitro group activating a nucleophilic aromatic substitution of a suitable leaving group X.
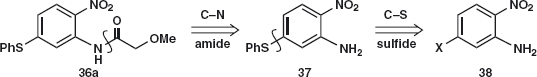
As it happens, the chloro-compound 38;X = Cl is available, though it could easily be made by nitration of meta-chloro-aniline 39. Displacement of chloride with the anion of PhSH gives 37. Acylation with methoxyacetyl chloride and reduction of the nitro group gives febantel.6

References
1. Clayden, Organic Chemistry, chapter 52, Polymerization.
2. T. Sohda, K. Mizuno, E. Imayima, Y. Sugiyama, T. Fujita and Y. Kawamatsu, Chem. Pharm. Bull., 1982, 30, 3580.
3. D. W. Robertson, J. H. Krushinski, R. W. Fuller and J. D. Leander, J. Med. Chem., 1988, 31, 1412.
4. M. Srebnik, P. V. Ramachandran and H. C. Brown, J. Org. Chem., 1988, 53, 2916.
5. Y. Gao and K. B. Sharpless, J. Org. Chem., 1988, 53, 4081.
6. Drug Synthesis, 4, 35.