

Contents
All books published by Wiley-VCH are carefully produced. Nevertheless, authors, editors, and publisher do not warrant the information contained in these books, including this book, to be free of errors. Readers are advised to keep in mind that statements, data, illustrations, procedural details or other items may inadvertently be inaccurate.
Library of Congress Card No.: applied for
British Library Cataloguing-in-Publication Data
A catalogue record for this book is available from the British Library.
Bibliographic information published by the Deutsche Nationalbibliothek
The Deutsche Nationalbibliothek lists this publication in the Deutsche Nationalbibliografie; detailed bibliographic data are available on the Internet at .
© 2011 WILEY-VCH Verlag & Co. KGaA, Boschstr. 12, 69469 Weinheim, Germany
All rights reserved (including those of translation into other languages). No part of this book may be reproduced in any form - by photoprinting, microfilm, or any other means - nor transmitted or translated into a machine language without written permission from the publishers. Registered names, trademarks, etc. used in this book, even when not specifically marked as such, are not to be considered unprotected by law.
Typesetting Laserwords Private Limited, Chennai, India
Printing and Binding Fabulous Printers Pte. Ltd., Singapore
Cover Design Formgeber, Eppelheim
List of Contributors
Bo Albinsson
Chalmers University of Technology Department of Chemical and Biological Engineering/Physical and Organic Chemistry Kemigarden 4 41296 Göteborg Sweden
David Q. Andrews
Environmental Working Group Washington, D.C. 20009 USA
Trisha L. Andrew
Massachusetts Institute of Technology Department of Chemistry 77 Massachusetts Avenue Cambridge, MA 02139 USA
Timothy Clark
Friedrich-Alexander-UniversitätErlangen-Nürnberg Department of Chemistry and Pharmacy & Interdisciplinary Center for Molecular Materials (ICMM) Egerlandstr. 3 91058 Erlangen Germany
Seong Ho Choi
University of Minnesota Department of Chemistry and Department of Chemical Engineering and Materials Science 421, Washinton Ave. SE Minneapolis, MN 55455 USA
Andrew R. Cook
Brookhaven National Laboratory Chemistry Department Upton, NY 11973 USA
Mattias P. Eng
Chalmers University of Technology Department of Chemical and Biological Engineering/Physical and Organic Chemistry Kemigarden 4 41296 Göteborg Sweden
C. Daniel Frisbie
University of Minnesota Department of Chemistry and Department of Chemical Engineering and Materials Science 421, Washinton Ave. SE Minneapolis, MN 55455 USA
Ferdinand C. Grozema
Delft University of Technology Department of Chemical Engineering Optoelectronic Materials Section Julianalaan 136 2628 BL Delft The Netherlands
Dirk M. Guldi
Friedrich-Alexander-UniversitätErlangen-Nürnberg Department of Chemistry and Pharmacy & Interdisciplinary Center for Molecular Materials (ICMM) Egerlandstr. 3 91058 Erlangen Germany
Magnus Hultell
Linköping University Department of Physics Chemistry and Biology IFM Bldg F Room G405 58183 Linköping Sweden
Frederick D. Lewis
Argonne/Northwestern Solar Energy Research (ANSER) Center Department of Chemistry 2145 Sheridan Road Evanston, IL 60208–3113 USA
Jerker Mårtensson
Chalmers University of Technology Department of Chemical and Biological Engineering/Physical and Organic Chemistry Kemigarden 4 41296 Göteborg Sweden
Nazario Martın
Universidad Complutense Departamento de Química Orgánica Facultad de Química 28040 Madrid Spain
John R. Miller
Brookhaven National Laboratory Chemistry Department Upton, NY 11973 USA
Mark A. Ratner
Argonne/Northwestern Solar Energy Research (ANSER) Center Department of Chemistry 2145 Sheridan Road Evanston, IL 60208–3113 USA
Kirk S. Schanze
University of Florida Chemistry Department Gainesville, FL 32611 USA
Laurens D. A. Siebbeles
Delft University of Technology Department of Chemical Engineering Optoelectronic Materials Section Julianalaan 136 2628 BL Delft The Netherlands
Gemma C. Solomon
Nano-Science Center and Department of Chemistry University of Copenhagen Universitetparken 5 Copenhagen Ø, 2100 Denmark
Paiboon Sreearunothai
Brookhaven National Laboratory Chemistry Department Upton, NY 11973 USA
and
Thammasat University Sirindhorn International Institute of Technology Pathum Thani, 12121 Thailand
Sven Stafström
Linköping University Department of Physics Chemistry and Biology IFM Bldg F Room G405 58183 Linköping Sweden
Timothy M. Swager
Massachusetts Institute of Technology Department of Chemistry 77 Massachusetts Avenue Cambridge, MA 02139 USA
Josh Vura-Weis
Argonne/Northwestern Solar Energy Research (ANSER) Center Department of Chemistry 2145 Sheridan Road Evanston, IL 60208–3113 USA
Michael R. Wasielewski
Argonne/Northwestern Solar Energy Research (ANSER) Center Department of Chemistry 2145 Sheridan Road Evanston, IL 60208–3113 USA
Mateusz Wielopolski
Friedrich-Alexander-UniversitätErlangen-Nürnberg Department of Chemistry and Pharmacy & Interdisciplinary Center for Molecular Materials (ICMM) Egerlandstr. 3 91058 Erlangen Germany
1
Introduction: Molecular Electronics and Molecular Wires
1.1 Introduction
According to the predictions of Gordon Moore in 1965, the number of transistors per square centimeter of silicon doubles every 18 months [1]. This requires that the size of transistors and the interconnecting wires between them decrease at the same rate. Up until now, this miniaturization has been realized by improvements in photolithographic techniques. These techniques will reach their fundamental limit in the near future, as the dimensions of the components drop below tens of nanometers. Therefore, it is of considerable practical and fundamental interest to study the smallest components that are likely to be functional, that is, components consisting of single molecules or groups of molecules.
Already in 1959 the eminent physicist Richard Feynman discussed the possibilities of devices of extremely small dimensions in his lecture entitled ‘‘There’s plenty of room at the bottom’’ [2]:
I don’t know how to do this on a small scale in a practical way, but I do know that computing machines are very large; they fill rooms. Why can’t we make them very small, make them of little wires, little elements - and by little I mean little. For instance, the wires should be 10 or 100 atoms in diameter, and the circuits should be a few thousand angstroms across. […] There is plenty of room to make them smaller. There is nothing that I can see in the laws of physics that says the computer elements cannot be made enormously smaller than they are now.
In 1959, Feynman and the rest of the world did not know how to manipulate electronic components on a molecular scale; however, more than 30 years after that, in the 1990s several breakthroughs were achieved and now, 50 years later, a large community of scientists is working on the use of single molecules as electronic components. Among the pioneers in single-molecule conduction studies were Gimzewski and Joachim who measured the electrical conductance of a single fullerene C60 molecule [3]. Other seminal experimental advances were the measurement of the electrical resistance of a single benzenedithiol bonded between two Au electrodes by the group of Reed et al. [4] and the experimental demonstration of single-molecule rectification in an Aviram-Ratner type molecule by Metzger and coworkers [5]. Since the 1990s, a lot of progress has been made, both in the practical problem of manipulating single molecules and doing measurements on them and in the fundamental understanding of the electrical processes on this small scale. As a result of this research, a variety of single-molecule electronic components have been proposed and demonstrated.
A field, that is, very much related to molecular electronics, and has inspired it to some extent, is that of electron transfer in donor-acceptor systems. This area of science started long before the first ideas of using molecules in electronics with the work of Mulliken in the late 1940s, from which the theory of binding and charge transfer spectra emerged [6]. A theory for electron transfer with a classical description of nuclear degrees of freedom was developed in the 1950s by Marcus [7–9] and later extended by Hush [10, 11]. Jortner and others later extended this theory by including a quantum mechanical description of the nuclear degrees of freedom [12, 13]. These theoretical predictions were confirmed experimentally over the following decades by (among many others) Verhoeven [14, 15], Paddon-Row [16, 17], and Miller [18, 19]. Most of the initial groundbreaking experiments were done for donor-bridge-acceptor systems in which the bridge consisted of a nonconjugated rigid spacer, most notably the norbornyl derivatives. These donor-bridge-acceptor molecules show strong resemblance to the initial molecular diode proposed by Aviram and Ratner [20]. More recently the study of electron transfer has been extended to conjugated bridges, with particular focus on the properties of conjugated chains as molecular wires [21–27].
In this chapter, we will not give a thorough review of the enormous progress that has been made in the field of single-molecule conductance. Excellent reviews on molecular electronics are available for a deeper background [28–35]. We aim to give an impression of some of the different molecular electronic components and discuss the importance of molecular wires that should serve as interconnects between these devices. We also discuss the different approaches that are used for studying charge transport through molecular wires. These approaches, both theoretically and experimentally, vary considerably between the fields of molecular electronics where conductance measurements are most common, and electron transfer where charge transfer is often determined by spectroscopic techniques. In the following chapters in this book, these different methods are discussed in detail and applied to actual systems.
1.2 Single-Molecule Devices
1.2.1 Molecular Rectifiers
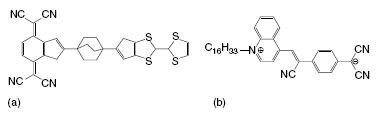
The first concrete idea for an electronic component consisting of a single molecule was the molecular rectifier described by Aviram and Ratner [20]. The molecular rectifier that they considered consisted of an electron donating moiety, tetrathiafulvalene, which was connected to an electron-accepting group, tetracyanoquinodimethane, by an ‘‘insulating’’ σ -bonded spacer, see Figure . This molecule can be considered as an analog of p-n junctions common to the design of traditional solid-state rectifiers. Quantum chemical calculations suggested that this molecule should indeed exhibit rectifying behavior. After this landmark proposal, it took another 25 years until such behavior was experimentally confirmed for the related donor-acceptor molecule shown in Figure by Metzger et al. [5, 36, 37].
Single-molecule transistors. (a) Aviram-Ratner proposal for a single-molecule rectifier. (b) Molecular rectifier realized by Metzger.
A more recent approach to realize a single-molecule rectifier, reported by the group of Dekker [38], is more akin to its macroscopic equivalent. It consists of single-walled nanotubes that can be either metallic or semiconducting depending on their diameter and helicity. An intramolecular junction between a metallic and a semiconducting nanotube section can be realized by introducing a pentagon and a heptagon into the hexagonal carbon lattice. Electrical transport measurements on a single carbon nanotube intramolecular metal-semiconductor junction have been performed [38]. It was shown that the transport characteristics were strongly asymmetric with respect to the bias polarity, thus exhibiting the behavior of a rectifying diode. The disadvantage of using carbon nanotubes is that there is no synthetic control over the construction of the molecules and the realization relies on coincidence during the synthesis of carbon nanotubes.
1.2.2 Molecular Switches
The basic control element in electronic architecture is the switch, which allows the control of current flow. Switches can be used in isolated form but can also be connected in arrays of multiple switches to implement logic operations [39, 40]. One example of a switch on a molecular scale is the photochromic switch consisting of a dithienylethene molecule; see Figure [41]. The connection between the thienyl rings can be opened or closed by illuminating with different wavelengths of light. In the open form, the thienylene rings are not connected and, therefore, the conjugation across the molecule is broken. If the molecule is illuminated with ultra-violet (UV) light, the closed form is obtained. The molecule can be switched back to its open form by irradiation with visible light. Such a light switchable molecule can be used as a memory element, using the open and closed form as ‘‘on’’ and ‘‘off’’ bits. The photochromic switch can also, in principle, be used for switching currents ‘‘on’’ or ‘‘off’’ on a molecular level when it is incorporated into a molecular wire or, as has been shown recently, by trapping it between two metal electrodes. The operation of this switch was demonstrated by chemisorbing it inside a mechanically controllable break junction between two gold electrodes. It was found that the resistance increased by 3 orders of magnitude upon opening of the switch by irradiation with visible light [42].
Single-moleculeswitches.(a)DithienyletheneswitchthatcanbeopenedandclosedbyilluminationwithvisibleandUVlight,respectively. (b)Redoxswitchthatcanbemadeconductingbyreductionoftheanthraquinonemoiety.
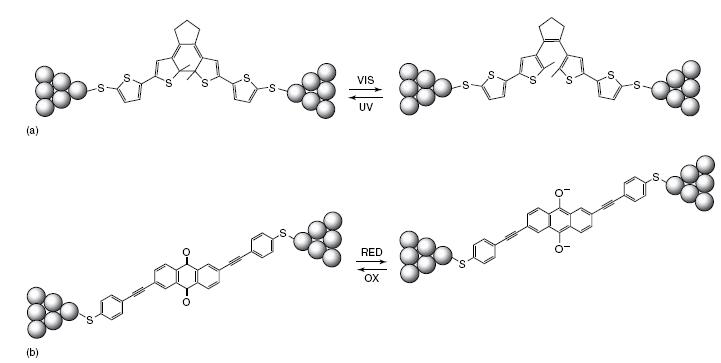
Another example is the anthraquinone-based switch reported by the group of Hummelen; see Figure [43]. In this molecule, the π-electron pathway can be switched from cross-conjugated to conjugated by reduction of the anthraquinone moiety. In general, charge transfer through a cross-conjugated π-system is much less efficient than charge transfer through a conjugated pathway [44, 45]. Therefore this molecule can be considered a redox switch.
1.2.3 Molecular Transistors
The examples of single-molecule switches discussed above rely on conformational changes in the molecule. This limits the possible switching speed to a few kilohertz since usually the reverse conformational transition is relatively slow in a molecular system [46]. An approach that should in principle allow much faster switching speeds is a switch (or transistor) that relies on a single electron transfer. In 1988, 14 years after the proposal of the molecular rectifier, Aviram proposed a field-effect transistor that consists of a single molecule; see Figure [47]. This transistor consists of a semiconducting piece of polythiophene, connected in such a way to a doped (oxidized) piece of polythiophene that charge transfer between these two parts of the molecule is inefficient. The oxidized polythiophene is conducting and the nondoped polythiophene will be nonconducting up to a certain threshold voltage, but the application of an electric field can result in tunneling of an electron between the two parts. In this way, the conduction of both polythiophene channels can be switched by application of an electric field [47].
Single-molecule transistors. (a) Aviram proposal for a molecular transistor consisting of a photoconductor coupled to a conductor. (b) A central ‘‘quantum dot’’ unit connected to three electrodes by conjugated chains.
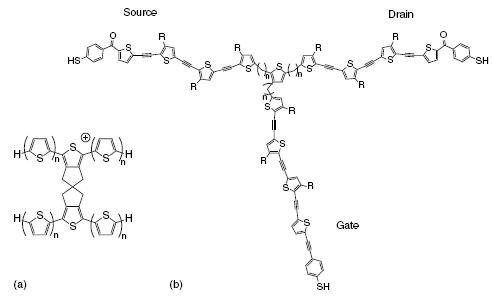
Another example of a single-molecule transistor that relies on single-electron tunneling is shown in Figure . In this three-terminal design, described by Wada [46, 48], a central ‘‘quantum dot’’ unit consisting of a single thiopene ring is connected to three conjugated arms by saturated linker units. In the case when two arms are connected to electrodes, the central part with the saturated linkages acts as a tunneling barrier. The tunneling rate through this barrier can be modified by applying a potential to the third terminal, resulting in an increase or decrease in the energy levels in the quantum dot part. Therefore, by applying a potential to the ‘‘gate’’ electrode, the tunneling current between the source and drain can be controlled. It has been estimated that switching speeds of more than 10 THz could be reached [49].
Single-molecule transistors that consist of a single semiconducting single-walled nanotube have been proposed by the group of Dekker [50]. The nanotubes are positioned across two Pt electrodes on a silicon oxide substrate with doped silicon as the back gate. The current through the nanotube can be manipulated by changing the voltage applied at the gate electrode. It has also been demonstrated that these devices can be assembled into one-, two-, and three-transistor circuits that perform a range of digital logic operations such as an inverter or a memory cell [51].
1.2.4 Molecular Wires: Connecting the Devices
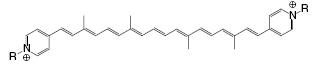
In order to use the single-molecule electronic components described above in a functional way while preserving the small scale, they have to be connected by conducting wires of the same (molecular) dimensions. One of the first to coin the term ‘‘molecular wire’’ was the 1988 Nobel prize winner Lehn who described a caroviologen molecule that could be incorporated into vesicle membranes, see Figure [52]. The charge in such a chain can transfer easily through the conjugated pathway between the two terminal groups of the molecule.
Caroviologen proposed by Lehn et al. as a trans-membrane molecular wire.
Similar conjugated molecular wires are the ‘‘simple’’ conjugated polymer-derived wires shown in Figures -(c). These wires consist of a piece of conjugated polymer analogous to the polymers used for organic electronics. In such conjugated polymers, generally there is a considerable amount of conformational freedom, most notably the rotational freedom around the (formally) single bonds in the chain [53]. Therefore, more rigid alternatives have also been proposed as show in Figure (d) [54]. One of the advantages of using organic molecules as molecular wires is the level of control over the structural and electronic properties of these wires. The conjugated wires can be designed to meet the required rigidity as, for example, in porphyrin-based molecular wires, see Figures (e) and (f). Butadiyne-linked porphyrin wires have interesting charge transfer properties but also exhibit a considerable degree of torsional disorder [25, 55, 56].
Examples of conjugated molecular wires,
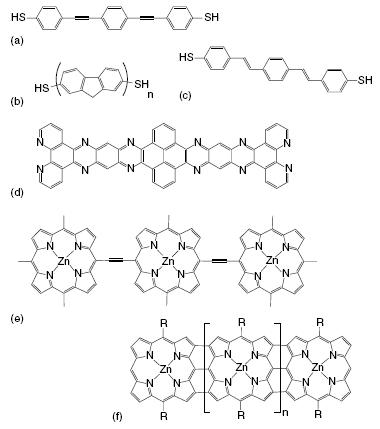
ranging from simple conjugated wires (a-c) to fully rigid oligo(quinoxaline) (d) and nonrigid (e) and rigid (f) porphyrin-based molecular wires.
These porphyrin wires can be made much more rigid by directly coupling the porphyrin units in a ladder-type structure as has been shown by Osuka et al. These porphyrin ‘‘tapes’’ should function as exceptionally efficient pathways for charge transport [57].
Apart from the synthetic control over the structure and properties inside a molecule, organic materials also offer advantages due to their self-assembling properties. Conjugated molecules can be designed so that they self-assemble into supra-molecular structures suitable for charge transport [58]. An excellent example of this is the incorporation of specific quadruple hydrogen-bonding units in conjugated molecules as shown by Meijer and coworkers [59–61]. Such designed self-assembly can possibly be used to assemble molecular devices and wires into electronic circuits that perform a specific function. It has already been shown that supramolecular interactions can be used to control the optical and charge transport properties of conjugated molecular wires [62–64].
1.3 Transport of Charges and Excitons in Molecular Wires
In the context of the emergence of molecular electronics, the study of charge transport through molecular wires has become an important research topic. Charge transport phenomena have been studied using a variety of techniques. These techniques can largely be divided in three categories. In the first category, the molecules are positioned between electrodes in some way [65, 66]. Sometimes single molecules are trapped between electrodes but often their properties are also studied in so-called self-assembled monolayers. In the latter case, the substrate functions as one electrode, while a scanning-tunneling microscopy tip is the other [35]. An example of such measurements is described in Chapter of this book.
The second approach to measuring the charge transport in conjugated molecular wires comes from the area of photo-induced electron transfer [21, 22]. In this case, an electron donor and acceptor are attached to a conjugated bridge and the rate of charge transfer upon excitation is measured by time-resolved spectroscopy (Figure c and d). This is extensively discussed in Chapters and for conjugated bridges and in Chapter for π-stacked DNA bridges.
Examples of conjugated molecular wires between electrodes (a, b) and in donor-bridge-acceptor systems (c, d).
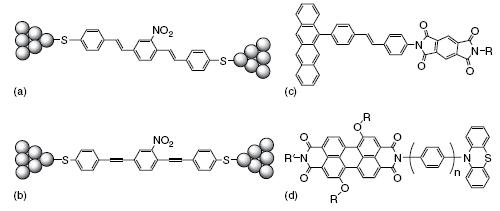
The basic mechanism of charge transfer involved in these two methods is very similar, even if the experimental methods differ considerably. In both cases, charge transfer is generally due to a single-step tunneling process in which a charge, either a hole or electron, tunnels between the donor and acceptor or between the electrodes without becoming localized on the bridge. In donor-bridge-acceptor systems, the rate of charge transfer can then be described in terms of the Marcus-Hush model, which involves coupling to the vibrational states in the molecule and its surroundings. It is then typically found that the rate of charge transfer decays exponentially with distance, since the charge transfer integral exhibits an exponential distance dependence [67, 68].
In the theoretical description of charge transport through molecules between electrodes, the Landauer approach has been used [69], see, for instance, Chapter of this book. Although the two approaches may appear very different at a first glance, they are very much related. The relation between the Landauer approach [69] for molecular conductance and the Marcus charge transfer rate has been demonstrated by Nitzan [70]. Similar to the case of single-step electron transfer, the conductance through the molecule typically decays exponentially with increasing distance between the electrodes. The groups of Joachim and Grill demonstrated this in 2009 in an experiment using a scanning tunneling microscope (STM). With the STM tip, a conjugated polymer was lifted off a conducting surface, while the current between the tip and surface was measured at the same time. Lifting the polymer off the surface increases the length of the conjugated chain between surface and tip, leading to an exponential decay of the conductance [71].
1.3.1 Deviations from Exponential Distance Dependence: Transfer to Hopping
In both the single-molecule conductance approach and spectroscopic measurements on donor-bridge-acceptor systems, interesting deviations from the exponential distance dependence have been observed. For donor-bridge-acceptor systems, Wasielewski and coworkers have found that after a certain bridge-length in conjugated molecules, the distance dependence of charge transfer becomes much weaker and is in fact nonexponential [22, 23]. The same trend has been observed for charge transfer through π-stacked DNA bases by the groups of Giese [72] and Lewis et al. [73, 74].
Interestingly, in single-molecule conductance experiments very similar observations were reported by the group of Frisbie for a series of conjugated chains of increasing length. Although the conductance was exponential for short chains, at a certain length the decrease with distance became much weaker, see also Chapter [75].
The fact that in both types of experiments the same deviations were observed confirms the strong similarity in the charge transfer process that is probed by the two approaches. The crossover from a strong exponential distance dependence to almost distance independence has in both cases been sought in a change of the mechanism by which the charge transfers. In the exponential regime, charge transfer occurs via a single-step (super-exchange) tunneling mechanism in which the charge is never localized on the bridge. For longer bridges, the superexchange tunneling process is very slow and actual population of the bridge by the charge becomes a competing process. In such cases, the charge can transfer from donor to acceptor or between electrodes by a multistep hopping mechanism. Theories describing the crossover between tunneling and incoherent hopping have been postulated [76–78]. In this respect, it is interesting to note that the presence of the charge on the bridge has not been observed experimentally, although in the case of charge transfer in DNA it was found that the charge leaves the donor faster than it arrives at the acceptor site [73, 74]. This indicates that the charge is at least temporarily localized on the bridge.
1.3.2 Charges Localized on Conjugated Chains
In both the spectroscopic studies on donor-bridge-acceptor systems and single-molecule conductance the charge does not become localized on the bridge in the majority of cases. As a consequence the rate of charge transfer or the conductance is determined to a large extent by the properties of the donor and acceptor or the molecule-electrode coupling. This means that, for instance, the charge transfer rate does not provide direct information on the motion of the charge when it is moving on the conjugated bridge. A convenient way of studying charges that are actually moving on conjugated chains is to generate the charges initially on the conjugated chains. This is possible by creating ionizations by irradiation with short pulses of high-energy electrons [79–82]. The charges generated in this way can move along conjugated polymer chains. This motion can be probed by optical spectroscopy, for instance, detecting the motion of charges toward appended traps at the chain ends, see Chapter [79]. Alternatively, it is possible to directly determine the mobility of the charges along the chains using the time-resolved microwave conductivity technique, as described in Chapter [80–82]. The dynamics of charges on conjugated polymers chains has also been studied theoretically as discussed in Chapters and .
The interesting feature of both these ways of probing charge transport is that the charge is actually localized on the chain and the motion along the chain is probed. It is hard to compare the data obtained from such measurements directly to the results from single-molecule conductance experiments or spectroscopy on donor-bridge-acceptor systems. However, in the limit of very long bridges, there should be similarities. In this limit, the single-step tunneling is by definition negligible and the only pathway for transport would be motion along the chain. Although it is experimentally quite challenging to go to this limit because of the small charge transfer rates (or low currents), it is of considerable fundamental interest to enter the regime where charges are moving on the chains.
1.3.3 Motion of Excited States
Although much of the focus in molecular electronics is on charge transport and electronic functionality, the molecules that are considered often also have interesting optical properties. In fact the combination of light and charges is one of considerable interest and, as illustrated above, in principle a current can be switched on and off by illumination of molecular switches with light of different wavelengths. In this context, excited states and, in particular, the motion of excited states along the molecular wires are of interest [83, 84]. Among the possible applications is the possibility to construct chemical sensors based on specific interactions that regulate motion of excited states [85]. The motion of excitons along conjugated chains is related to charge migration, and some of the techniques to probe the motion are the same. For instance, the motion of excitons to appended traps at the ends of conjugated chains is very similar to the trapping of charges on these traps. This is discussed in Chapter . Other ways of probing exciton motion along conjugated chains use the specific properties, for instance, fluorescence. In Chapter the depolarization of the fluorescence is used as a probe of the motion of excited states along conjugated chains.
References
1. Moore, G.E. (1965) Cramming more components onto integrated circuits. Electronics, 38, 114–117.
2. Feynman, R.P. (1960) There’s plenty of room at the bottom. Eng. Sci., 23, 22–36.
3. Joachim, C., Gimzewski, J.K., Schlitter, P.R., and Chavy, C. (1995) Electronic transparence of a single C60 molecule. Phys. Rev. Lett., 74, 2102–2105.
4. Reed, M.A., Zhou, C., Muller, C.J., Burgin, T.P., and Tour, J.M. (1997) Conductance of a molecular junction. Science, 278, 252–254.
5. Metzger, R.M., Chen, B., Hopfner, U., Lakshmikantham, M.V., Vuillaume, D., Kawai, T., Wu, X., Tachibana, H., Hughes, T.V., Sakurai, H., Baldwin, J.W., Hosch, C., Cava, M.P., Brehmer, L., and Ashwell, G.J. (1997) Unimolecular electrical rectification in hexadecylquinolinium tricyanoquinodimethnide. J. Am. Chem. Soc., 119, 10455- 10466.
6. Mulliken, R.S. (1952) Molecular compounds and their spectra II. J. Am. Chem. Soc., 74, 811–824.
7. Marcus, R.A. (1956) On the theory of oxidation-reduction reactions involving electron transfer. J. Chem. Phys., 24, 966–978.
8. Marcus, R.A. and Sutin, N. (1985) Elec-tron transfers in chemistry and biology. Biochim. Biophys. Acta, 811, 265–322.
9. Marcus, R.A. (1964) Chemical and electrochemical electron-transfer theory. Annu. Rev. Phys. Chem., 15, 155–196.
10. Hush, N.S. (1967) Intervalence-transfer absorption. Part 2. Theoretical considerations and spectroscopic data. Prog. Inorg. Chem., 8, 391.
11. Hush, N.S. (1968) Homogeneous and heterogeneous optical and thermal electron transfer. Electrochim. Acta, 13, 1005–1023.
12. Ulstrup, J. and Jortner, J. (1976) The effect of intramolecular quantum modes on free energy relationships for electron transfer reactions. J. Chem. Phys., 63, 4358–4368.
13. Bixon, M. and Jortner, J. (1999) Electron transfer – from isolated molecules to biomolecules. Adv. Chem. Phys., 106, 35–202.
14. Oevering, H., Paddon-Row, M.N., Heppener, M., Oliver, A.M., Cotsaris, E., Verhoeven, J.W., and Hush, J.W. (1987) Long-range photoinduced through-bond electron transfer and radiative recombination via rigid nonconjugated bridges: distance and solvent dependence. J. Am. Chem. Soc., 109, 3258–3269.
15. Warman, J.M., de Haas, M.P., Paddon-Row, M.N., Cotsaris, E., Hush, N.S., Oevering, H., and Verhoeven, J.W. (1986) Light-induced giant dipoles in simple model compounds for photosynthesis. Nature, 320, 615–616.
16. Hush, N.S., Paddon-Row, M.N., Cotsaris, E., Oevering, H., Verhoeven, J.W., and Heppener, M. (1985) Distance dependence of photoinduced electron transfer through non-conjugated bridges. Chem. Phys. Lett., 117, 8–11.
17. Paddon-Row, M.N. (1994) Investigating long-range electron-transfer processes with rigid covalently linked donor-(norbornylogous bridge)-acceptor systems. Acc. Chem. Res., 27, 18–25.
18. Closs, G.L. and Miller, J.R. (1988) Intramolecular long-distance electron transfer in organic molecules. Science, 240, 440–447.
19. Penfield, K.W., Miller, J.R., Paddon-Row, M.N., Cotsaris, E., Oliver, A.M., and Hush, N.S. (1987) Optical and thermal electron transfer in rigid difunctional molecules of fixed distance and orientation. J. Am. Chem. Soc., 109, 5061–5065.
20. Aviram, A. and Ratner, M.A. (1974) Molecular rectifiers. Chem. Phys. Lett., 29, 277–283.
21. Davis, W.B., Svec, W.A., Ratner, M.A., and Wasielewski, M.R. (1998) Molecular-wire behavior in p-phenylenevinylene oligomers. Nature, 396, 60–63.
22. Weiss, E.A., Ahrens, M.J., Sinks, L.E., Gusev, A.V., Ratner, M.A., and Wasielewski, M.R. (2004) Making a molecular wire: charge and spin transport through para-phenylene oligomers. J. Am. Chem. Soc., 126, 5577–5584.
23. Goldsmith, R.H., Sinks, L.E., Kelley, R.F., Betzen, L.J., Liu, W., Weiss, E.A., Ratner, M.A., and Wasielewski, M.R. (2005) Wire-like charge transport at near constant bridge energy through fluorene oligomers. Proc. Natl. Acad. Sci. U.S.A., 102, 3540–3545.
24. Wiberg, J., Guo, L., Pettersson, K., Nilsson, D., Ljungdahl, T., Martensson, J., and Albinsson, B. (2006) Charge recombination versus charge separation in donor–bridge–acceptor systems. J. Am. Chem. Soc., 129, 155–163.
25. Winters, M.U., Dahlstedt, E., Blades, H.E., Wilson, C.J., Frampton, M.J., Anderson, H.L., and Albinsson, B. (2007) Probing the efficiency of electron transfer through porphyrin-based molecular wires. J. Am. Chem. Soc., 129, 4291–4297.
26. Kils  , K., Kajanus, J., Macpherson, A.N., Mrtensson, J., and Albinsson, B. (2001) Bridge dependent electron transfer in porphyrin-based donor–bridge–acceptor systems. J. Am. Chem. Soc., 123, 3069–3080.
, K., Kajanus, J., Macpherson, A.N., Mrtensson, J., and Albinsson, B. (2001) Bridge dependent electron transfer in porphyrin-based donor–bridge–acceptor systems. J. Am. Chem. Soc., 123, 3069–3080.
27. Giacalone, F., Segura, J.L., Martin, N., and Guldi, D.M. (2004) Exceptionally small attenuation factors in molecular wires. J. Am. Chem. Soc., 126, 5340–5341.
28. Muller, C.J. and Reed, M.A. (1996) There is plenty of room between two atom contacts. Science, 272, 1901–1902. 29. Joachim, C., Gimzewski, J.K., and Aviram, A. (2000) Electronics using hybrid-molecular and mono-molecular devices. Nature, 408, 541–548.
30. Carroll, R.L. and Gorman, C.B. (2002) The genesis of molecular electronics. Angew. Chem. Int. Ed. Engl., 41, 4378–4400.
31. Heath, J.R. and Ratner, M.A. (2003) Molecular electronics. Phys. Today, 5, 43–49.
32. McCreery, R.L. (2004) Molecular electronic junctions. Chem. Mater., 16, 4477- 4496.
33.Heath, J.R. (2009) Molecular electronics. Annu. Rev. Mater. Res., 39, 1–23.
34.Moth-Poulsen, K. and Bjornholm, T. (2009) Molecular electronics with single molecules in solid-state devices. Nat. Nanotechnol., 4, 551–556.
35.McCreery, R.L. and Berggren, A.J. (2009) Progress with molecular electronic junctions: meeting experimental challenges in design and fabrication. Adv. Mater., 21, 4303–4322.
36.Metzger, R.M. (2001) The quest for unimolecular rectification from Oxfordto Waltham to Exeter to Tuscaloosa. J. Macromol. Sci., A38, 1499- 1517.
37.Metzger, R.M. (2003) Unimolecular electrical rectifiers. Chem. Rev., 103, 3803- 3834.
38.Yao, Z., Postma, H.W.C., Balents, L., and Dekker, C. (1999) Carbon nanotube intramolecular junctions. Nature, 402, 273–276.
39.Irie, M. (2000) Diarylethenes for memories and switches. Chem. Rev., 100, 1685- 1716.
40.Feringa, B.L. (2001) Molecular Switches, Wiley-VCH Verlag GmbH, Weinheim.
41.Kudernac, T., Katsonis, N., Browne, W.R., and Feringa, B.L. (2009) Nano-electronic switches: light-induced switching of the conductance of molecular system. J. Mater. Chem., 19, 7168- 7177.
42.Dulic, D., van der Molen, S.J., Kudernac, T., Jonkman, H.T., de Jong, J.J.D., Bowden, T.N., van Esch, J., Feringa, B.L., and van Wees, B.J. (2003) Phys. Rev. Lett., 91, 207402.
43.Van Dijk, E.H., Myles, D.J.T., Van der Veen, M.H., and Hummelen, J.C. (2006) Synthesis and properties of an anthraquinone-based redox switch for molecular electronics. Org. Lett., 8, 2333- 2336.
44.Mayor, M., Weber, H.B., Reichert, J., Elbing, M., Von Hanisch, C., Beckmann, D., and Fischer, M. (2003) Electric current through a molecular rod – relevance of the position of the anchor groups. Angew. Chem. Int. Ed. Engl., 42, 5834- 5838.
45.Van der Veen, M.H., Rispens, M.T., Jonkman, H.T., and Hummelen, J.C. (2004) Molecules with linear pi-conjugated pathways between all substituents: omniconjugation. Adv. Funct. Mater., 14, 215–223.
46.Wada, Y. (1999) A prospect for single molecule information processing devices. Pure Appl. Chem., 71, 2055- 2066.
47.Aviram, A. (1988) Molecules for memory, logic and amplification. J. Am. Chem. Soc., 110, 5687–5692.
48.Wada, Y. (1999) Proposal of atom/molecule switching devices. J. Vac. Sci. Technol. A, 17, 1399- 1405.
49.Lutwyche, M.I. and Wada, Y. (1994) Estimate of the ultimate performance of the single-electron transistor. J. Appl. Phys., 74, 3654–3661.
50.Tans, S.J., Verschueren, A.R.M., and Dekker, C. (1998) Room-temperature transistor based on a single carbon nanotube. Nature, 393, 49–52.
51.Bachtold, A., Hadley, P., Nakanishi, T., and Dekker, C. (2001) Logic circuits with carbon nanotube transistors. Science, 294, 1317- 1320.
52.Arrhenius, T.S., Blanchard-Desce, M., Dvolaitzky, M., and Lehn, J.-M. (1986) Molecular devices: caroviologens as an approach to molecular wires-synthesis and incorporation into vesicle membranes. Proc. Natl. Acad. Sci. USA, 83, 5355- 5359.
53.Grozema, F.C., van Duijnen, P.T., Berlin, Y.A., Ratner, M.A., and Siebbeles, L.D.A. (2002) Intramolecular charge transport along isolated chains of conjugated polymers: effect of torsional disorder and polymerization defects. J. Phys. Chem. B, 106, 7791–7795.
54.Ishow, E., Gourdon, A., Launay, J.-P., Chiorboli, C., and Scandola, F. (1999) Synthesis, mass spectrometry, and spectroscopic properties of a dinuclear ruthenium complex comprising a 20 A long fully aromatic bridging ligand. Inorg. Chem., 38, 1504–1510.
55.Winters, M.U., Karnbratt, J., Eng, M., Wilson, C.J., Anderson, H.L., and Albinsson, B. (2007) Photophysics of a butadiyne-linked porphyrin dimer: influence of conformational flexibility in de ground and first singlet excited state. J. Phys. Chem. C, 111, 7192–7199.
56.Kocherzhenko, A.A., Patwardhan, S., Grozema, F.C., Anderson, H.L., and Siebbeles, L.D.A. (2009) Mechanism of charge transport along zinc porphyrin-based molecular wires. J. Am. Chem. Soc., 131, 5522–5529.
57.Kim, D. and Osuka, A. (2004) Directly linked porphyrin arrays with tunable excitonic interactions. Acc. Chem. Res., 37, 735–745.
58.Hoeben, F.J.M., Jonkheijm, P., Meijer, E.W., and Schenning, A.P.H.J. (2005) About supramolecular assemblies of pi-conjugated systems. Chem. Rev., 105, 1491–1546.
59.Sijbesma, R.P., Beijer, F.H., Brunsveld, L., Folmer, B.J.B., Hirschberg, J.H.K.K., Lange, R.F.M., Lowe, J.K.L., and Meijer, E.W. (1997) Reversible polymers formed from self-complementary monomers using quadruple hydrogen bonding. Science, 278, 1601- 1604.
60.El-ghayoury, A., Schenning, A.P.H.J., Van Hal, P.A., Van Duren, J.K.J., Janssen, R.A.J., and Meijer, E.W. (2001) Supramolecular hydrogen-bonded OPVS polymers. Angew. Chem. Int. Ed. Engl., 40, 3660–3663.
61.Schenning, A.P.H.J., Jonkheim, P., Peeters, E., and Meijer, E.W. (2001) Hierarchical order in supramolecular assemblies of hydrogen bonded oligo (p-phenylene vinylenes). J. Am. Chem. Soc., 123, 409–416.
62.Grozema, F.C., Houarner-Rassin, C., Prins, P., Siebbeles, L.D.A., and Anderson, H.L. (2007) Supramolecular control of charge transport in molecular wires. J. Am. Chem. Soc., 129, 13370- 13371.
63.Frampton, M.J. and Anderson, H.L. (2007) Insulated molecular wires. Angew. Chem. Int. Ed. Engl., 46, 1028–1064.
64.Cacilli, F., Wilson, J.S., Michels, J.J., Daniel, C., Silva, C., Friend, R.H., Severin, N., Samori, P., Rabe, J.P., O’Connell, M.J., Taylor, P.N., and Anderson, H.L. (2002) Cyclodextrin-threaded conjugated polyrotaxanes as insulated molecular wires with reduced interstrand interactions. Nat. Mater., 1, 160–164.
65.James, D.K. and Tour, J.M. (2005) Molecular wires. Top. Curr. Chem., 257, 33–62.
66.Love, J.C., Estroff, L.A., Kriebel, J.K., Nuzzo, R.G., and Whitesides, G.M. (2005) Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem. Rev., 105, 1103–1169.
67.Edwards, P.P., Gray, H.B., Lodge, M.T.J., and Williams, R.J.P. (2008) Electron transfer and electronic conduction through and intervening medium. Angew. Chem. Int. Ed. Engl., 47, 6758–6765.
68.Berlin, Y.A., Grozema, F.C., Siebbeles, L.D.A., and Ratner, M.A. (2008) Charge transfer in donor-bridge-acceptor systems: static disorder, dynamic fluctuations and complex kinetics. J. Phys. Chem. C, 112, 10988- 11000.
69.Landauer, R. (1957) Spatial variation of currents and fields due to localized scatterers in metallic conduction. IBM J. Res. Dev., 1, 223–231.
70.Nitzan, A. (2001) A relationship between electron-transfer rates and molecular conduction. J. Phys. Chem. A, 105, 2677–2679.
71.Lafferentz, L., Ample, F., Yu, H., Hecht, S., Joachim, C., and Grill, L. (2009) Conductance of a single conjugated polymer as a continuous function of its length. Science, 323, 1193–1197.
72.Giese, B., Amaudrut, J., Kohler, A.-K., Spormann, M., and Wessely, S. (2001) Direct observation of hole transfer through DNA by hopping between adenine bases and by tunneling. Nature, 412, 318–320.
73.Lewis, F.D., Zhu, H., Daublain, P., Cohen, B., and Wasielewski, M.R. (2006) Hole mobility in DNA A tracts. Angew. Chem. Int. Ed. Engl., 45, 7982–7985. 74.Lewis, F.D., Zhu, H., Daublain, P., Fiebig, T., Raytchev, M., Wang, Q., and Shafirovich, V. (2006) Crossover from superexchange to hopping as the mechanism for photo-induced charge transfer in DNA hairpin conjugates. J. Am. Chem. Soc., 128, 791–800.
75.Choi, S.H., Kim, B., and Frisbie, C.D. (2008) Electrical resistance of long conjugated molecular wires. Science, 320, 1482- 1486.
76.Bixon, M. and Jortner, J. (2002) Long-range and very long range charge transport in DNA. Chem. Phys., 281, 393–408.
77.Berlin, Y.A., Burin, A.L., and Ratner, M.A. (2002) Elementary steps for charge transport in DNA: thermal activation vs. tunneling. Chem. Phys., 275, 61–74.
78.Giese, B. (2000) Long-distance charge transport in DNA: the hopping mechanism. Acc. Chem. Res., 33, 631–636.
79.Asaoka, S., Takeda, N., Iyoda, T., Cook, A.R., and Miller, J.R. (2008) Electron and hole transport to trap groups at the ends of conjugated polyfluorenes. J. Am. Chem. Soc., 130, 11912- 11920.
80.Hoofman, R.J.O.M., de Haas, M.P., Siebbeles, L.D.A., and Warman, J.M. (1998) Highly mobile electrons and holes on isolated chains of the semiconducting polymer poly(phenylenevinylene). Nature, 392, 54–56.
81.Grozema, F.C., Hoofman, R.J.O.M., Candeias, L.P., de Haas, M.P., Warman, J.M., and Siebbeles, L.D.A. (2003) The formation and recombination kinetics of positively charged poly(phenylene vinylene) chains in pulse-irradiated dilute solutions. J. Phys. Chem. A, 107, 5976- 5986.
82.Grozema, F.C., Siebbeles, L.D.A., Warman, J.M., Seki, S., Tagawa, S., and Scherf, U. (2002) Hole conduction along molecular wires: sigma-bonded silicon versus pi-bond-conjugated carbon. Adv. Mater., 14, 228–231.
83.Hennebicq, E., Pourtois, G., Scholes, G.D., Herz, L.M., Russell, D.M., Silva, C., Setayesh, S., Grimsdale, A.C., Mullen, K., Bredas, J.-L., and Beljonne, D. (2005) Exciton migration in rigid-rod conjugated polymers: an improved Forster model. J. Am. Chem. Soc., 127, 4744- 4762.
84.Dykstra, T.E., Hennebicq, E., Beljonne, D., Gierschner, J., Claudio, G., Bittner, E.R., Knoester, J., and Scholes, G.D. (2009) Conformational disorder and ultra-fast exciton relaxation in PPV-family conjugated polymers. J.Phys. Chem. B, 113, 656–667.
85.Thomas, S.W. III, Joly, G.D., and Swager, T.M. (2007) Chemical sensors based on amplifying fluorescent conjugated polymers. Chem. Rev., 107, 1339- 1389.
Part I
Molecules between Electrodes