
Inhaltsverzeichnis
Vorwort
Adressen der Autoren
1 Definition, historischer Abriß und Bedeutung der Photochemie
2 Die konzeptionellen und theoretischen Grundlagen der Photochemie (W.-D. Stohrer)
2.1 Die Natur der elektromagnetischen Strahlung
2.2 Die photochemische Reaktion, eine Wanderung auf und zwischen Potentialflächen
2.3 Orbitale, Konfigurationen und Zustände
2.4 Die Lichtabsorption und -emission
2.5 Der strahlungslose Wechsel zwischen Potentialflächen
2.6 Die Desaktivierung eines durch Lichtabsorption gebildeten elektronisch angeregten Zustandes
2.7 Der mögliche Ablauf photochemischer Reaktionen
3 Photoreaktionen organischer Verbindungen (M. Tausch)
3.1 Tabellarische Übersicht
3.2 Photolysen organischer Moleküle
3.3 Photoadditionen
3.4 Photoisomerisierungen
3.5 Photoreduktionen, Photooxidationen
3.6 Literatur zu Kapitel 3
4 Photochemie im sichtbaren Bereich, solare Photochemie und verwandte Prozesse (D. Wöhrle)
4.1 Der sichtbare Bereich im elektromagnetischen Spektrum und Farbe
4.2 Künstliche Lichtquellen und solare Einstrahlung
4.3 Photosynthese
4.4 Lösungsprozesse
4.5 Photochromie
4.6 Literatur zu Kapitel 4
5 Photochemie und Photophysik in selbstorganisierenden Systemen, hochmolekularen Verbindungen und Festkörpern (D. Wöhrle)
5.1 Photochemie in selbstorganisierenden Systemen
5.2 Photochemie in organischen und anorganischen hochmolekularen Verbindungen
5.3 Photochemische Polymerisation und Photopolymerisation
5.4 Anorganische und organische Halbleiter
5.5 Literatur zu Kapitel 5
6 Chemolumineszenz (H. Brandl)
6.1 Einleitung und Begriffsbestimmung
6.2 Chemolumineszenz-Systeme
6.3 Literatur zu Kapitel 6
7 Photochemie in Technik, Biologie und Medizin
7.1 Photochemie in der Technik und möglicher Anwendung (D. Wöhrle)
7.2 Der Photoreaktor Atmosphäre (M. Tausch)
7.3 Photochemie und Biologie (M. Tausch, D. Wöhrle)
7.4 Photochemie/Photophysik und Medizin (D. Wöhrle)
7.5 Photochemie, alkoholische Getränke und ausgiebiges Sonnenbaden (D. Wöhrle)
7.6 Literatur zu Kapitel 7
8 Arbeitsmethoden und Versuche
8.1 Arbeitsmethoden zur Durchführung photochemischer Experimente (D. Wöhrle)
8.2 Instrumenten analytische Methoden (D. Wöhrle)
8.3 Quantifizierung photochemischer Reaktionen und photophysikalischer Prozesse (D. Wöhrle)
8.4 Versuche zur Photochemie und Photophysik (M. Tausch, D. Wöhrle)
8.5 Versuche zur Chemolumineszenz (H. Brandl)
8.6 Allgemeine Literatur zu Kapitel 8
9 Anhang (D. Wöhrle)
9.1 Angaben über Einheiten, Umrechnungsfaktoren, Größenordnungen
9.2 Glossar zu Definitionen in der Photochemie
9.3 Weitere Literatur zur Photochemie
Index
Lehrbücher für fortgeschrittene Studenten von WILEY-VCH
L. H. Gade
Koordinationschemie
1998, ISBN 3-527-29503-8
T. Linker, M. Schmittel
Radikale und Radikalionen in der Organischen Synthese
1998, ISBN 3-527-29492-9
E. L. Eliel, S. H. Wilen
Organische Stereochemie
Übersetzung herausgegeben von H. Hopf und J. Mulzer
1998, ISBN 3-527-29349-3
J. A. Gewert, J. Görlitzer, S. Götze, J. Looft, P. Menningen, T. Nobel, H. Schirok, C. Wulff
Problems!
Ein Übungsbuch zur organischen Synthese
1998, ISBN 3-527-29516-X

Prof. Dr. Dieter Wöhrle
Institut für Organische und Makromolekulare Chemie
Universität Bremen, NW 2
Postfach 33 04 40
D-28334 Bremen
Prof. Dr. Michael W. Tausch
Institut für Synthesechemie
Gerhard-Mercator-Universität
GH Duisburg
Lotharstraße 1
D-47057 Duisburg
Prof. Dr. Wolf-Dieter Stohrer
Institut für Organische Chemie
Universität Bremen, NW 2
Postfach 33 04 40
D-28334 Bremen
Titelbild:
Wir danken dem Aulis Verlag Deubner & Co KG, Köln, und dem C. C. Buchner Verlag, Bamberg, für die freundliche Abdruckgenehmigung der Bilder auf dem Buchumschlag.
Die Deutsche Bibliothek – CIP-Einheitsaufnahme
Wöhrle, Dieter:
Photochemie : Konzepte, Methoden, Experimente / Dieter Wöhrle ;
Michael W. Tausch ; Wolf-Dieter Stohrer. Unter Mitarb. von Herbert Brandl. –
Weinheim ; New York ; Chichester ; Brisbane ; Singapore ; Toronto :
Wiley-VCH, 1998
ISBN 3-527-29545-3
© WILEY-VCH Verlag GmbH, D-69469 Weinheim (Federal Republic of Germany), 1998
Allc Rechte, insbesondere die der Übersetzung in andere Sprachen, vorbehalten. Kein Teil dieses Buches darf ohne schriftliche Genehmigung des Verlages in irgendeiner Form – durch Photokopie, Mikroverfilmung oder irgendein anderes Verfahren – reproduziert oder in eine von Maschinen, insbesondere von Datenverarbeitungsmaschinen, verwendbare Sprache übertragen oder übersetzt werden. Die Wiedergabe von Warenbezeichnungen, Handelsnamen oder sonstigen Kennzeichen in diesem Buch berechtigt nicht zu der Annahme, daß diese von jedermann frei benutzt werden dürfen. Vielmehr kann es sich auch dann um eingetragene Warenzeichen oder sonstige gesetzlich geschützte Kennzeichen handeln, wenn sie nicht eigens als solche markiert sind.
All rights reserved (including those of translation into other languages). No part of this book may be reproduced in any form – by photoprinting, microfilm, or any other means – nor transmitted or translated into a machine language without written permission from the publishers. Registered names, trademarks, etc. used in this book, even when not specifically marked as such, are not to be considered unprotected by law.
Print ISBN 9783527295456
Epdf ISBN 978-3-527-66107-7
Epub ISBN 978-3-527-66088-9
Mobi ISBN 978-3-527-66087-2
Vorwort
Die Photochemie ist ein Paradebeispiel für Interdisziplinarität. Photochemie beinhaltet in erster Linie die Beschäftigung mit und die Anwendung von Anorganischer, Organischer, Physikalischer und Theoretischer Chemie bei Vorgängen der Einwirkung von Licht auf Materie. Aber auch für benachbarte Disziplinen der Chemie wie Physik, Biologie und Medizin liefert die Photochemie entscheidende Grundlagen der Wechselwirkung zwischen Licht und Materie. Das vorliegende Lehrbuch trägt diesem interdisziplinären Charakter Rechnung. Die Intention dieses Werkes ist, eine Lücke unter den Lehrbüchern zu schließen: Es handelt sich um einen Einführungslehrgang, der sich in erster Linie an Studierende der Chemie und benachbarter Disziplinen wendet, aber auch an andere in der wissenschaftlichen Praxis Tätige, für deren Arbeitsgebiete die Wechselwirkung zwischen Licht und Materie von Bedeutung ist. Die Auseinandersetzung mit dem Buch soll für eine vertiefende theoretische und experimentelle Bearbeitung photochemischer Inhalte Anreize geben und Voraussetzungen schaffen.
Kapitel 1 bringt einen kurzen Rückblick auf einige historische Meilensteine in der Entwicklung der Photochemie und führt über knappe Definitionen in die aktuelle Photochemie und ihre Bedeutung ein. Darauf aufbauend werden im Kapitel 2 die konzeptionellen Grundlagen photochemischer Prozesse entwickelt. Dabei geht es weniger um mathematisierte Quantenmechanik, sondern vielmehr um die anwendungsorientierte Erläuterung der für die Photochemie wichtigsten quantenchemischen Prinzipien und Modelle, die auch nicht unbedingt streng erklärt, sondern häufig nur plausibel, sprich verständlich, “begreifbar” gemacht werden sollen. Deshalb werden - wo immer möglich und notfalls auch auf Kosten der Puristik - halbklassische oder klassische Betrachtungsweisen mit einbezogen.
Das Kapitel 3 arbeitet die wichtigsten organischen Photoreaktionen auf. Dem inzwischen sehr umfangreichen Faktenmaterial auf diesem Gebiet wird durch einen tabellarischen Überblick zu Beginn des Kapitels Rechnung getragen. Bei der Darstellung einzelner Reaktionstypen wird nicht in erster Linie Vollständigkeit angestrebt, sondern das grundlegende Verständnis und die systematische Einteilung organischer Photoreaktionen. Einzelne Beispiele können anhand der im vorletzten Kapitel angegebenen Versuche vertieft werden.
Die nächsten vier Kapitel beinhalten wichtige Teilbereiche der Photochemie und verwandter Gebiete. Dazu wird zunächst im Kapitel 4 die Photochemie mit sichtbarem Licht vorgestellt. Hier stehen neben photochemischen und photochromen Reaktionen auch die solare Einstrahlung und als Musterprozeß im sichtbaren Bereich die Photosynthese im Vordergrund.
Im Kapitel 5 werden verschiedene Prozesse der Wechselwirkung Licht-Materie zusammengeführt. Das sind zum einen photochemische Prozesse in Wirt-Gast-Sytemen, in Kombination mit hochmolekularen Verbindungen bzw. Initiierung photochemischer Polymerisationen. Zum anderen werden aber auch Prozesse an anorganischen Halbleitern in photovoltaischen, photoelektrochemischen Zellen bzw. in Photosensibilisierungszellen und an Halbleiterpartikeln behandelt.
Das Kapitel 6 widmet sich dem interessanten Phänomen der Chemolumineszenz. Dazu werden die wichtigsten chemischen Reaktionen, die unter Abgabe von Licht ablaufen, vorgestellt. Die Behandlung dieses Themenbereiches umfaßt auch den gegenwärtigen Stand des mechanistischen Ablaufs dieser Reaktionen.
Den Autoren ist es wichtig, auch anwendungsorientierte Inhalte einzubringen. Dazu stehen im Kapitel 7 beispielhaft photochemische Prozesse in Industrie und weiteren Anwendungen im Vordergrund. Auch werden neben der Atmosphärenchemie beispielhaft Prozesse und Anwendungen in Biologie und Medizin behandelt.
Das Kapitel 8 ist nun ganz den Arbeitsmethoden und Versuchen gewidmet. Dabei werden zunächst die wichtigsten Inhalte zur Laborpraxis, der Anwendung von Methoden der Absorption bzw. Aussagen zur Kinetik und Energetik photochemischer Reaktionen gebracht. Besonders im Vordergrund stehen dann Versuche zur Photochemie und Chemolumineszenz. Die Versuche können teilweise für studienbegleitende Praktika adaptiert oder für Experimentalvorlesungen verwertet werden. Bei der Anfertigung mancher Diplomarbeit oder einer Dissertation möge dieser Teil des Buches mit Versuchsideen und/oder konkreten Experimentieranleitungen hilfreich sein.
Den Abschluß des Buches bildet das Kapitel 9 mit Angaben über Daten und Definitionen der Photochemie und verwandter Bereiche.
Es versteht sich von selbst, daß nicht alle Teilbereiche der Photochemie und verwandter Gebiete eingehend behandelt werden können. Soweit dies nicht der Fall ist, sind aber Literaturhinweise angegeben. Die Autoren hoffen, daß neben Chemikern auch Physiker, Biologen und Mediziner, die sich mit der Wechselwirkung von Licht und Materie befassen, durch dieses Buch profitieren können.
Besonderer Dank gilt denjenigen, die in unterschiedlicher Art an dem Buch mitgewirkt haben. Hier sprechen wir in erster Linie Herrn Studienrat Herbert Brandlunsere Anerkennung aus, der engagiert das Kapitel und Versuche über Chemolumineszenz eingebracht hat. Weiterhin danken wir den Autoren, die mit Versuchsvorschriften beigetragen haben. Besonders dankbar sind wir Frau Rita Fofana, Frau Ulrike Labudda, Herrn Dr. Günter Schnurpfeil und Herrn Dipl. Chem. Tobias Borrmann für die aufwendige Hilfe bei der redaktionellen Erstellung des Manuskriptes bis zum fertigen Layout.
Bremen und Duisburg, im Frühjahr 1998
D. Wöhrle, M. W. Tausch, W.-D. Stohrer
Adressen der Autoren
Wolf-Dieter Stohrer, Institut für Organische Chemie, Universität Bremen, NW 2, Postfach 330 440, 28334 Bremen, Deutschland; Tel. 0421-218 2953.
Michael W. Tausch, Institutfür Synthesechemie, Gerhard-Mercator-Universität, GH Duisburg, Lotharstr. 1, 47057 Duisburg, Deutschland; Tel. 0203-379 2207, Fax 0203-379 2540.
Dieter Wöhrle, Institut für Organische und Makromolekulare Chemie, Universität Bremen, NW 2, Postfach 330 440, 28334 Bremen, Deutschland; Tel. 0421-218 2805/2809, Fax 0421-218 4935.
Unter Mitarbeit von:
Herbert Brandl, Bettina-von-Arnim-Str. 8, 24568 Kaltenkirchen; Tel. 04191-8392.
Beiträge zu den Versuchen im Kapitel 8.4:
Vincenzo Augugliaro, Vittorio Loddo, Leonardo Palmisano, Mario Schiavello,Dipartimento di Ingegneria Chimica dei Processi e dei Materiali, University of Palermo, Viale delle Scienze,90128 Palermo, Italien(Versuch 43).
T. Bach, Fachbereich Chemie der Philipps-Universität Marburg, Hans-Meerweinstr., 35043 Marburg, Deutschland(Versuch 9).
D. Döpp, Institut für Synthesechemie, Gerhard-Mercator-Universität, GH Duisburg, Lotharstr. 1, 47057 Duisburg, Deutschland (Versuch 30).
Heinz Dürr, Fachrichtung Organische Chemie, Universität des Saarlandes, Bau 23.2, 66041 Saarbrücken, Deutschland (Versuch 28).
Michael Grätzel, Institut für Physikalische Chemie, Swiss Federal Institute of Technology, 1015 Lausanne, Schweiz; G. Smestad, Paul-Scherrer-Institut 104-1 5232, Schweiz (Versuch 35).
Andreas Hartwig, Andreas Härder, Fraunhofer-Institut für Angewandte Materialforschung, Bereich Klebtechnik und Polymere, Neuer Steindamm 2, 28719 Bremen, Deutschland (Versuche 18,19).
Barbara Heller, Günther Oehme, Institut für Organische Katalyseforschung an der Universität Rostock e.V., D-18055 Rostock, Buchbinderstr. 5/6, Deutschland (Versuch 7).
M. Kaneko, Faculty of Science, Ibaraki-University, Bunyko, Mito, 310 Japan (Versuche 27, 41,42).
H. Kisch, Institut für Anorganische Chemie, Universität Erlangen - Nürnberg, Egerlandstr.1, 91058 Erlangen, Deutschland (Versuch 31).
Jochen Mattay, Björn Schlummer, Institut für Organische Chemie, Universität Kiel, Olshausenstr. 40, 24098 Kiel; Ernst-Ulrich Würthwein, Organisch-Chemisches Institut, Universität Münster, Corrensstr.40, 48149 Münster, Deutschland (Versuche 3, 4,24).
MiguelA. Miranda,H. Garcia und M.L.Cano, Departamento de Quimica/Instituto de Tecnología Química UPV-CSIC, Universidad Politecnica de Valencia, Apartado 22012, 46071 Valencia, Spanien(Versuch 8).
C.A. Mitsopoulou, D. Katakis, Inorganic Chemistry Laboratory, University of Athens, Panepistimiopolis, 15771 Athen, Griechenland; E. Vrachnou, N.C.S.R. “Demokritos”, P.O.Box 60228, 15310 Aghia Paraskevi, Attiki, Griechenland (Versuch 29).
A.V. Nikolaitchik, Center for Photochemical Sciences, Bowling Green State University, Bowling Green, Ohio 43403, USA (Versuch 37).
H.-D. Scharf, P. Esser, Institut für Organische Chemie der Technischen Hochschule, Professor-Pirlet-Straße 1, 52056 Aachen, Deutschland (Versuche 20,22).
Als photochemische Reaktionen werden im landläufigen (engeren) Sinne allgemein die Reaktionen bezeichnet, bei denen die für die Reaktion notwendige Aktivierungsenergie nicht in Form von Wärme, sondern in Form von sichtbarem oder ultraviolettem Licht1 zugeführt wird, die Reaktion also nicht durch Bunsenbrenner oder Heizpilz, sondern durch Sonne oder künstliche Stahlungsquellen initiiert wird. Im allgemeineren Sinne versteht man aber unter photochemischen Reaktionen diejenigen, die nicht ausschließlich - wie dies bei den thermischen Reaktionen der Fall ist - im elektronischen Grundzustand ablaufen, sondern bei denen entlang der Reaktionskoordinate auch ein oder mehrere elektronisch angeregte Zustände involviert sind. Dies hat zur Folge, daß wir neben den lichtinduzierten photochemischen Reaktionen im engeren Sinne auch weitere Prozesse betrachten werden, und zwar jeweils unter theoretisch-fachsystematischen, experimentellen und anwendungsbezogenen Gesichtspunkten. Zum einen berücksichtigen wir die zu den lichtinduzierten komplementären lichtproduzierenden Reaktionen, die als Chemolumineszenz bezeichnet werden. Zum anderen sind Fluoreszenz und Phosphoreszenz, photovoltaische und photoelektrochemische Prozesse zu nennen, die sich eher mit physikalischen Prozessen elektronisch angeregter Zustände befassen.
Reaktionen, die durch Röntgen- oder γ-Strahlen initiiert werden, werden aus praktischen Gründen definitionsgemäß nicht der Photochemie, sondern der Radiochemie zugeordnet und bleiben im folgenden unberücksichtigt.
Auf unserem Planeten sind Photoreaktionen bedeutend älter als das Leben. Sie sind eng mit der Bildung organischer Moleküle in der präbiotischen Phase und dann mit der Evolution des Lebens verknüpft (Kap. 7.3.1.1). Die Photosynthese ist bis heute der zentrale Prozeß für Energie, Nahrung und Klima. Die ersten Kenntnisse des Menschen über Vorgänge mit Lichtbeteiligung verlieren sich in frühen Zeiten der Kulturgeschichte. Mit dem Licht ihres verehrten Sonnengottes präparierten die alten Ägypter vor 4500 Jahren die Mumien ihrer Pharaonen, und mit gebündeltem Sonnenlicht zündete vor 2200 Jahren der Grieche Archimedes die Segel der feindlichen Schiffe an. Alexander der Große benutzte wohl die Farbveränderungen eines photochromen Farbstoffs als erste photochemische Reaktion, um den Angriff seiner Truppen zu koordinieren (s. Beginn des Kap. 4.5). In der biblischen Schöpfungsgeschichte heißt es im Ersten Buch Moses, Vers 3-4: „Und Gott sprach: Es werde Licht! / Und es ward Licht. / Und Gott sah, daß das Licht gut war. “ Es ist bemerkenswert, daß bereits die Schreiber des Alten Testaments im Licht eine der Urschöpfungen erkannten, die zu den Voraussetzungen für die Entstehung irdischen Lebens gehören.
Lange Zeit stand die thermische Nutzung der Sonnenenergie bei chemischen Reaktionen im Vordergrund. So beschrieb Libavius 1608 in seinem berühmten Werk „Alchymia“ mehrere Methoden für die Fokussierung von Sonnenlicht auf bestimmte Flächen und die Nutzung für chemische Veränderungen [1]. Doch das Phänomen Licht blieb über Jahrtausende hinweg in eine Aura von Mystik und Magie gehüllt, noch mehr als das Feuer, an das es untrennbar gebunden schien. Diese Vorstellung fiel, als Brand, der Hamburger Alchimist, im Jahr 1669 das kalte Licht des Calcinationsrückstandes aus menschlichem Urin entdeckte. Brands Licht war - entgegen der Meinung einiger Zeitgenossen - weder der Stein der Weisen, noch das Elixier des Feuers. Er hatte das Element Phosphor erhalten, das zwar nie die Alchimistenträume von der Goldmacherei erfüllte, aber u.a. zur Erkenntnis verhalf, daß von Menschenhand erzeugtes Licht nicht immer von Feuer oder heißen Körpern ausgehen mußte. Es handelt sich bei Brands Entdeckung um das älteste überlieferte Beispiel von Chemolumineszenz (vgl. Kap. 6).
Etwa ein Jahrhundert nach der Entdeckung des Phosphors, als Lavoisier in minutiöser 13-jähriger Arbeit (1772 bis 1785) die Phlogistontheorie der Verbrennung Punkt für Punkt entkräftet und durch die Sauerstofftheorie ersetzt hatte, stand bereits längst fest, daß das Leuchten des weißen Phosphors eine Begleiterscheinung seiner Oxidation ist. Und wieder ein knappes Jahrhundert später, im Jahre 1862, beobachtete Edmund Bequerel, dessen Sohn Henri 34 Jahre später die natürliche Radioaktivität entdecken sollte, daß kaltes Licht auch von einigen Körpern erzeugt werden kann, wenn sie kurz vorher mit Licht bestrahlt worden waren. Da aber hierbei keine stofflichen Änderungen erfolgen, wurde dieses Phänomen, die Phosphoreszenz, ebenso wie die damit verwandte Fluoreszenz, zunächst nicht zum näheren Forschungsobjekt der Chemie. Der als Begründer der Elektrochemie bekannte Physikochemiker J. W. Ritter beobachtete um 1800, daß Silbersalze auch jenseits der violetten Farbe aus dem Spektrum geschwärzt werden. Daher gilt er als Entdecker des UV-Lichtes. Er stellte in der damals üblichen, romantisch-schwärmerischen Ausdrucksweise fest “Licht ist die Quelle jeglicher Kraft, die Leben schafft und Thätigkeit.”
J. Priestley beobachete um 1790 zwei „echte“ Photoreaktionen. Er setze Ampullen gefüllt mit dem „spirit of nitre“ (Salpetersäure) dem Sonnenlicht aus und beobachtete eine rötliche Verfärbung. Dies muß als erste Photoreaktion in der Gasphase angesehen werden. Auch erarbeitete Priestley erste Ergebnisse zur Photosynthese (s. Kap. 4.3). Das 1817 erschienene „Handbuch der theoretischen Chemie“ von L. Gmelin stellt eine gute Übersicht zu den Kenntnissen und Theorien über das Licht Anfang des 19. Jahrhunderts dar [2], In 26 Punkten werden die Wirkungen des Lichtes auf „wägbare Stoffe“ wie die Chlorknallgasreaktion, die Reaktionen des Chlorwassers (Arbeiten u.a. von C. L. Berthollet, 1790), die Schwärzung des Silberchlorides, die Zerstörung von Pflanzenfarben, die Entwicklung von Sauerstoff aus „Kohlensäure“ durch grüne Pflanzenteile beschrieben. In die Zeit von 1840 bis 1860 fallen weitere Arbeiten zur photolytischen Chlorknallgasreaktion (R. Bunsen und H. Roscoe bzw. J. W. Drapers und W. C. Wittwers [3]), zur chemischen Wirkung des Lichtes auf eine wäßrige Lösung von Eisen(III)oxid und Salzsäure - heute als Ferrioxalat-Aktinometrie bekannt - (J. W. Döbereiner) und zur Umlagerung von Santonin als wohl am längsten bekannte Lichtreaktion (s. Kap. 5; H, Trommsdorf u.a. [4]). Die Folgezeit von etwa 1875 bis 1900 war initiiert durch die Entwicklung der präparativen Chemie, geprägt von photochemischen Arbeiten zu Dimerisierungen (u.a. J. Fritzsche, C. T. Liebermann, J. Bertram und R. Kürsten), cis-trans-Isomerisierungen (u.a. W. H. Perkin, J. Wislicenus, C. T. Liebermann), aromatischen Halogenierungen (u.a. J. Schramm), Reduktion von Carbonylverbindungen (u.a. H. Klinger) und Reaktionen von Diazo- bzw. Diazoniumverbindungen (u.a. A. Feer) [4].
Seit Lavoisier, bis tief ins 20.Jahrhundert hinein, spielte die thermische Chemie in der chemischen Praxis eine weitaus größere Rolle als die Lichtchemie. Im Labor wie in der Industrie wurden fast alle Reaktionen durch Wärmezufuhr in Gang gesetzt und/oder in Gang gehalten; bei exothermen Reaktionen mußte, besonders in der Technik, Wärme abgeführt werden. Auch das chemische Denken wurde von der Energieform Wärme beherrscht. Sprach Lavoisier im Jahr 1789 noch vom „Kaloricum - der unwägbaren Materie der Wärme “, sollte sich Dalton etwa 20 Jahre später in seinem berühmten Werk „A New System of Chemical Philosophy” wie folgt äußern: „Jedes Teilchen (Atom) nimmt den Mittelpunkt einer verhältnismäßig großen Sphäre ein und behauptet seine Würde dadurch, daß es alle übrige, welche vermöge ihrer Schwere oder aus anderen Gründen geneigt wären, es aus seiner Stelle zu vertreiben, in einer ehrfurchtsvollen Entfernung hält… diese weit ausgedehnten Sphären bestehen aus Wärmestoff. “ So nimmt es kein Wunder, daß die Wärme namensgebend und Mittelpunkt der Thermodynamik, der einzigen chemischen Grundtheorie bis zur Quantenmechanik, war und ist.
Unter den lichtinduzierten Reaktionen hat sich die Photographie mit Silberhalogeniden bereits im 19. Jahrhundert als eigenständiger Zweig etabliert und ist das auch bis heute geblieben. Am Anfang des 20. Jahrhunderts bekam die präparative Photochemie dann weitere Impulse durch G. Ciamician und zwischen den Jahren 1937 und 1957 durch A. Schönberg. Im Licht der ägyptischen Sonne brachte er in Kairo zahlreiche Verbindungen zur Reaktion und faßte seine Ergebnisse in dem 1958 erschienenen und heute noch geschätzten Buch „Präparative organische Photochemie” zusammen [5]. In die Zeit von etwa 1928 bis 1950 fallen auch die Entdeckung weiterer Chemolumineszenz-Reaktionen (s. Kap. 6). Mit der lichtinduzierten Chlorierung von Alkanen und Aromaten zog die Photochemie auch in die chemische Großtechnik ein. Dennoch führte die Photochemie bis etwa 1950 eher ein Schattendasein.
Der Siegeszug der Quantenmechanik durch die theoretische Chemie des 20. Jahrhunderts bewirkte, daß die Beteiligung von elektromagnetischer Strahlung bei chemisch relevanten Prozessen zunehmend in den Vordergrund rückte. Das Tor zur Chemie der angeregten Zustände war entriegelt; eine Flut von theoretischen Voraussagen setzte ein; die Laborpraxis bestätigte in beeindruckender Weise ihre Richtigkeit. Zu den bekanntesten Beispielen dafür gehören die Woodward-Hoffmann-Regeln für pericyclische Reaktionen (Regeln von der Erhaltung der Orbitalsymmetrie) und die Salem-Korrelationsdiagramme.
Doch intellektuelle Faszination allein reicht häufig nicht aus, um die Forschung auf einem Gebiet rasch voranzutreiben, handfester praktischer Nutzen muß daran gekoppelt sein. Besonders seit den Siebziger und Achtziger Jahren des 20. Jahrhunderts ist das für die Photochemie der Fall. Auf der Anwendungsseite entwickelte sich ein breites Spektrum von spektroskopischen Methoden für chemische Analyse und Strukturaufklärung. Es reicht von der inzwischen fast archaischen UV/Vis-Spektroskopie bis zu den verschiedenen Spektroskopien mit monochromatischem und gepulstem Laserlicht. Quer durch alle Wellenlängenbereiche steht ein Angebot an Varianten für Emissions- und Absorptionsspektroskopien zur Verfügung. Die Superlative in der Spurenanalyse sind mittels Laser-Resonanzionisationsspektroskopie, Laser-RIS und Chemolumineszenz-Assay realisierbar. Man dringt beim Nachweis bestimmter Substanzen bis in den ppq-Bereich bzw. sogar bis in den Attomol-Bereich. Industriell allerdings konnten sich bislang die Photooxidation, die Photochlorierung, die Photosulfochlorierung und die Photonitrosierung von Kohlenwasserstoffen in größerem Maße durchsetzen (vgl. Tab. 7-1). Die Photochemie “in dünnen Schichten”, bei der Materialoberflächen durch Lichteinwirkung gezielt verändert werden, ist seit einem Jahrzehnt stark im Vormarsch (vgl. Kap. 7.1.2 - 7.1.4). In ihrer wirtschaftlichen Bedeutung hat sie bereits heute die Photochemie “im Kessel” überholt.
1. A. Libavius, Alchymia, 1608; nachgedruckt in: D. Diderot, Encyclopedie, 1766; vgl. Gmelin-Institut (Hrsg.), Die Alchemie des Andreas Libavius, VCH Verlagsgesellschaft, Weinheim, 1985.
2. L. Gmelin, Handbuch der theoretischen Chemie, Band 1, Frankfurt a.M., 1817; Nachdruck Weinheim, 1967.
3. U. Boberlin, Photochemische Untersuchungen von R. Bunsen und H. Roscoe im Vergleich mit den Arbeiten J. W. Drapers und W. C. Wittwers, Verlag Köster, Berlin, 1993.
4. H. D. Roth, Die Anfänge der organischen Photochemie, Angew. Chem. 1989, 111, 1220-1234.
5. A. Schönberg, Präparative organische Photochemie, Springer-Verlag, Berlin, 1958.
1 Im folgenden wird unter dem Begriff „Licht“ nicht nur das Licht im engeren Sinne, also die für unser Auge sichtbare elektromagnetische Strahlung verstanden, sondern allgemein die elektromagnetische Strahlung, die bei Absorption durch Atome oder Moleküle Elektronen aus deren energetisch höherliegenden besetzten Orbitalen in deren energetisch tieferliegenden unbesetzten Orbitale promoviert. Dieses ist durch elektromagnetische Strahlung im sichtbaren, im nahen und mittleren UV-Bereich und - sehr gelegentlich - im nahen IR-Bereich möglich.
Die Natur der elektromagnetischen Strahlung ist zwiespältig.
Schon Newton wollte das sichtbare Licht als aus kleinsten Teilchen bestehend verstanden wissen, wohl oder vielleicht in der Hoffnung, seine grundlegenden Gesetze der klassischen Mechanik auch auf das Licht anwenden zu können. Für Huygens hingegen, einen Zeitgenossen Newtons, war das Licht eine Welle im für den Menschen nicht wahrnehmbaren „Äther“, eine Vorstellung mit der er schon damals die Beugung des Lichts, ja sogar dessen Doppelbrechung am Kalkspatkristall einsichtig (auf heute noch akzeptierte Weise) erklären konnte; dennoch hielt sich die Newtonsche Korpuskulartheorie des Lichtes hartnäckig und hatte - vielleicht ob ihrer größeren Anschaulichkeit, vielleicht ob der wissenschaftlichen Autorität Newtons - eine große Anhängerschaft, wenngleich weder die Beugung des Lichtes, noch dessen Doppelbrechung am Kalkspatkristall, noch sonst irgend etwas damit erklärt werden konnte.
Erst die Untersuchungen Youngs und Fresnels Anfang des 19. Jahrhunderts zur Interferenz des Lichtes versetzten der Korpuskulartheorie den augenblicklichen und - scheinbar - endgültigen Todesstoß. Die ausschließliche Wellennatur des Lichtes sollte damit für die nächsten 100 Jahre völlig unbestritten gelten, um so mehr, nachdem Mitte des 19. Jahrhunderts der elektromagnetische Wellencharakter des Lichtes von Maxwell theoretisch verstanden und damit untermauert worden war1.
An der Wende vom 19. zum 20. Jahrhundert sah sich Planck bei seinen theoretischen Untersuchungen zur Strahlung schwarzer Körper zu der Annahme gezwungen, bei der Absorption und Emission elektromagnetischer Strahlung durch Materie finde die damit gekoppelte Energieumwandlung nicht kontinuierlich statt, wie dies die damals außer Frage stehende ausschließliche Wellennatur der elektromagnetischen Strahlung zwingend forderte, sondern diskontinuierlich in Form von einzelnen „Energiepaketen“; die Energie E eines derartigen diskreten Energiepakets sei proportional der Frequenz v der elektromagnetischen Strahlung, also E = hv, wobei die Proportionalitätskonstante h als Plancksches Wirkungsquantum (h = 6,626 10−34 J s) zu einer der wichtigsten Naturkonstanten werden sollte.
Planck hielt seine Annahme von den diskreten Energiepaketen für einen unbefriedigenden - weil unrealistischen - mathematischen Artefact, den er nolens volens fürs erste und - wie er hoffte - nur vorübergehend gebrauchen mußte, um die experimentellen Ergebnisse zur Strahlung des schwarzen Körpers analytisch geschlossen formulieren zu können.
1905 postulierte Einstein bei seiner durch den Nobelpreis 1921 gewürdigten Interpretation des photoelektrischen Effekts, die Planckschen Energiepakete - heute bekanntlich Photonen oder Lichtquanten genannt - seien kein mathematischer Artefact, sondern physikalische Realität, nicht nur beim Energieaustausch zwischen Materie und elektromagnetischer Strahlung, sondern auch beim Energietransport der elektromagnetischen Strahlung2.
Der Teilchen-Welle-Dualismus der elektromagnetischen Strahlung war geboren, eine posthume Genugtuung für Newton, wenngleich diese Lichtquanten keine materiellen Teilchen sind und nicht der Newtonschen Mechanik gehorchen, sondern der Quantenelektrodynamik.
Der heute allgemein akzeptierte Teilchen-Welle-Dualismus entzieht sich der Vorstellungskraft des Menschen als - im Sinne der Physik - makroskopischem Wesen. Dieser Teilchen-Welle-Dualismus kann deshalb nicht verstanden, „begriffen“ werden. Es muß einfach zur Kenntnis genommen werden, daß sich je nach Experiment der Teilchencharakter (z. B. photoelektrischer Effekt) oder der Wellencharakter (z. B. Interferenz) der elektromagnetischen Strahlung offenbart.
Analog der Kopenhagener Deutung der Quantenmechanik ist die Wahrscheinlichkeitsdichte der Lichtquanten - und damit die Energiedichte der elektromagnetischen Strahlung - zu einer bestimmten Zeit an einer bestimmten Stelle proportional der Intensität der elektromagnetischen Welle zu dieser Zeit an dieser Stelle. Damit läßt sich auch die Interferenz als typisches Kriterium des Wellencharakters der elektromagnetischen Welle mit dem Teilchencharakter der elektromagnetischen Welle in Einklang bringen. Die Brücke zwischen Teilchen- und Wellencharakter ist geschlagen!
Bei den Diskussionen der Wechselwirkungen zwischen Licht und Materie in den folgenden Kapiteln werden je nach Fragestellung sowohl der Teilchencharakter wie der Wellencharakter des Lichtes gleichermaßen herangezogen werden.
Tab. 2-1 zeigt die Energie der elektromagnetischen Strahlung als Funktion deren Farbe und Wellenlänge bzw. Frequenz.
Der Vergleich mit einigen typischen Bindungsenergien (C-C: 341 kJ mol−1; C-H: 413 kJ mol−1; H-H: 436 kJ mol−1; C-Cl: 328 kJ mol−1; C-Br: 276 kJ mol−1; F-F: 155 kJ mol−1; I-I: 151 kJ mol−1) zeigt, daß ein Photon von der Thermodynamik her sehr wohl in der Lage ist, eine chemische Bindung zu spalten. Ob aber ein zu einer Bindungsspaltung hinreichend energiereiches Photon von einem Molekül überhaupt absorbiert werden kann, und ob im Falle der Absorption die Energie des Photons auch zur Spaltung einer Bindung benützt wird, ist eine andere Frage; eine Frage, deren Beantwortung breiten und zentralen Raum in den folgenden Kapiteln einnehmen wird.
Tabelle 2-1. Energie der elektromagnetischen Strahlung (in kJ pro mol Photonen) als Funktion der Farbe und Wellenlänge (in nm) bzw. Frequenz (in s−1); vgl. auch Abb. 9-1

Da bei photochemischen Reaktionen nicht nur - wie bei den thermischen Reaktionen - der elektronische Grundzustand, sondern auch ein oder mehrere elektronisch angeregte Zustände involviert sind, ist es für das Verständnis photochemischer Reaktionen notwendig, die gemeinsamen wie die unterschiedlichen Eigenschaften der verschiedenen Elektronenzustände, und die Möglichkeiten und Ursachen von Übergängen zwischen diesen zu kennen und zu verstehen.
Ein Molekül aus m Kernen und n Elektronen im Zustand j wird in der Quantenmechanik durch eine Zustandsfunktion(2-1)
(2-1) 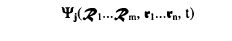
beschrieben. Diese Zustandsfunktion (2-1) ist eine Funktion der Kernkoordinaten  der Elektronenkoordinaten ri und der Zeit t, und ist Lösung der (zeitabhängigen) Schrödinger-Gleichung
der Elektronenkoordinaten ri und der Zeit t, und ist Lösung der (zeitabhängigen) Schrödinger-Gleichung 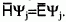
Befindet sich das Molekül in einem stationären Zustand3, dann kann die Zustandsfunktion (2-1) in das Produkt (2-2)
(2-2) 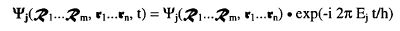
separiert werden, wobei  und Ej die Lösungen der zeitunabhängigen Schrödinger-Gleichung
und Ej die Lösungen der zeitunabhängigen Schrödinger-Gleichung  sind.
sind.
Der zeitunabhängige, nur von den Koordinaten der Kerne und Elektronen abhängige erste Faktor (2-3)
(2-3) 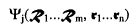
im Produkt (2-2) muß korrekterweise als die (nur ortsabhängige) Amplitudenfunktion der Zustandsfunktion (2-2) bezeichnet werden, was allerdings nur selten geschieht. Denn in der Praxis - und leider auch in vielen Lehrbüchern - wird auf die Wiedergabe des zeitabhängigen komplexen Exponentialtermes exp(−i 2π Ej t/h) in der Zustandsfunktion (2-2) häufig, ja fast immer verzichtet, und lediglich die Amplitudenfunktion (2-3) schlampig als die Zustandsfunktion des stationären Zustandes j bezeichnet. Bei der Bestimmung von Eigen- und Erwartungswerten eines stationären Zustandes spielt der zeitabhängige komplexe Exponentialterm in der Tat keine Rolle, da er entweder herausfällt (Erwartungswert), oder lediglich einen konstanten Faktor darstellt (Eigenwert). Aber die - insbesondere didaktisch-methodisch - nützlichen Analogien zwischen der quantenmechanischen Zustandsfunktion (2-2) eines stationären Zustandes einerseits, und der Funktion einer klassischen stehenden Welle andererseits können mit der Amplitudenfunktion (2-3 alleine nicht verstanden und angewandt werden; dazu bedarf es auch des zeitabhängigen Exponentialtermes. Erst recht gilt dies bei nichtstationären Zuständen, wie beispielsweise für den Übergang eines elektronisch angeregten Zustandes in den Grundzustand unter Emission eines Photons (vgl. Kap. 2.4.3).
Nach dieser Fest- und Klarstellung wird nun aber dennoch vereinbart, daß auch hier und im folgenden bei der Behandlung stationärer Probleme häufig - wenn immer ohne Mißverständnis möglich - lediglich die Amplitudenfunktion (2-3) als die Zustandsfunktion des (stationären) Zustandes j bezeichnet wird, wohl wissend, daß für die eigentliche Zustandsfunktion (2-2) noch der zeitabhängige komplexe Exponentialterm vonnöten ist, und gelegentlich auch vonnöten sein wird!
Die Amplitudenfunktion, oder - vereinbarungsgemäß einfacher - die Zustandsfunktion (2-3) hängt gleichzeitig vom „Verhalten“ der Elektronen und der Kerne ab. Das Verhalten der Elektronen hängt also auch von dem der Kerne ab, und umgekehrt. Aufgrund ihrer um mehrere Zehnerpotenzen geringeren Masse sind die Elektronen um mehrere Zehnerpotenzen weniger träge als die Kerne, d. h. einer Änderung der Kernanordnung und der dadurch bedingten Änderung der Coulombkräfte werden die Elektronen schnell folgen, einer Änderung der Elektronenanordnung und der dadurch bedingten Änderung der Coulombkräfte aber die Kerne nur langsam. Aus der „Sicht“ der Kerne sind die Elektronen schnell, fast unendlich schnell. Aus der „Sicht“ der Elektronen sind die Kerne langsam, fast unendlich langsam.
Die Born-Oppenheimer-Approximation streicht nun die beiden Wörter „fast“ und geht davon aus, daß sich die Elektronen einer Änderung der Kernanordnung unendlich rasch anpaßten, bzw. daß aus der Sicht der Elektronen die Kernbewegungen unendlich langsam seien, so daß für das momentane Verhalten der Elektronen die momentane Bewegung der Kerne keine Rolle spiele, sondern nur deren momentane Anordnung.
Mit dieser Annahme kann - ohne dies im einzelnen hier zu zeigen - die Funktion (2-3) ihrerseits in ein Produkt (2-4)
(2-4) 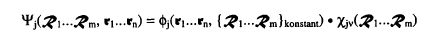
mit den Faktoren
(2-5) 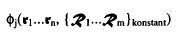
und
(2-6) 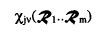
separiert werden.
Die Funktion (2-5), die als Variablen nur die Elektronenkoordinaten ri enthält, beschreibt die beobachtbaren Größen (Observablen) der Elektronen, also z. B. deren Aufenthaltswahrscheinlichkeit, im Elektronenzustand j unter dem Einfluß des Potentialfeldes des ruhend gedachten Kerngerüstes mit den festen Kernkoordinaten 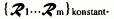
Die Funktion (2-6), die als Variablen nur die Kernkoordinaten enthält, beschreibt die Observablen der Kerne, also z. B. deren Aufenthaltswahrscheinlichkeit während der Kernschwingung im Kernschwingungszustandv des Elektronenzustandes
Die Funktion (2-5) beschreibt also das Verhalten der Elektronen im stationären Elektronenzustand j unter dem Einfluß des Kerngerüstes mit vorgegebenen, ruhend angenommenen Kernkoordinaten Diese Funktion  j ist Lösung der zeitunabhängigen Schrödinger-Gleichung:
j ist Lösung der zeitunabhängigen Schrödinger-Gleichung:
(2-7) 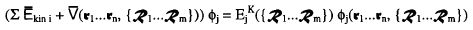
 ist der Operator der kinetischen Energie des Elektrons i,
ist der Operator der kinetischen Energie des Elektrons i,  ist der Operator der potentiellen Gesamtenergie von Elektronen und ruhenden Kernen, und
ist der Operator der potentiellen Gesamtenergie von Elektronen und ruhenden Kernen, und  ist die Gesamtenergie. Da der Hamiltonoperator
ist die Gesamtenergie. Da der Hamiltonoperator 
 auch die potentielle Energie der ruhenden Kerne berücksichtigt, enthält die Gesamtenergie nicht nur die kinetische und potentielle Energie der Elektronen im Elektronenzustand j unter dem Einfluß des ruhend gedachten Kernfeldes
auch die potentielle Energie der ruhenden Kerne berücksichtigt, enthält die Gesamtenergie nicht nur die kinetische und potentielle Energie der Elektronen im Elektronenzustand j unter dem Einfluß des ruhend gedachten Kernfeldes  sondern auch noch die potentielle Energie des Kerngerüstes. Diese Gesamtenergie hängt als Eigenwert der stationären Elektronenzustandsfunktion j natürlich nicht von den Koordinaten ri, der Elektronen ab, sondern nur von den Koordinaten des vorgegebenen ruhenden Kerngerüstes und kann deshalb aus der Sicht der Kerne als potentielle Energie verstanden werden.
sondern auch noch die potentielle Energie des Kerngerüstes. Diese Gesamtenergie hängt als Eigenwert der stationären Elektronenzustandsfunktion j natürlich nicht von den Koordinaten ri, der Elektronen ab, sondern nur von den Koordinaten des vorgegebenen ruhenden Kerngerüstes und kann deshalb aus der Sicht der Kerne als potentielle Energie verstanden werden.
Für jede mögliche feste Kernanordnung ergibt die Lösung der Schrödinger-Gleichung (2-7) jeweils die Energie  und die Zustandsfunktion (2-5) für jeden stationären Elektronenzustand j. Die Gesamtheit aller so berechneter Energien eines Elektronenzustandes j für alle möglichen Kernanordnungen ergibt (als „Ordinate“) gegen die 3m-6 Achsen der Kernkoordinaten (als „Abszissen“) aufgetragen die (3m-6+l)-dimensionale Energiehyperfläche - auch Potentialfläche genannt - dieses Elektronenzustandes j.
und die Zustandsfunktion (2-5) für jeden stationären Elektronenzustand j. Die Gesamtheit aller so berechneter Energien eines Elektronenzustandes j für alle möglichen Kernanordnungen ergibt (als „Ordinate“) gegen die 3m-6 Achsen der Kernkoordinaten (als „Abszissen“) aufgetragen die (3m-6+l)-dimensionale Energiehyperfläche - auch Potentialfläche genannt - dieses Elektronenzustandes j.
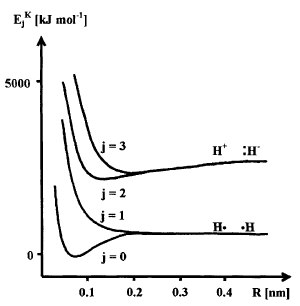
Dies wird in Abb. 2-1 am Beispiel des H2-Moleküls veranschaulicht, für das bei verschiedenen festen Kernabständen R jeweils die Energien 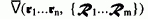 der vier energetisch tiefsten Elektronenzustände j = 0, 1, 2, 3 berechnet wurden, die dann die Potentialflächen, die sich hier natürlich auf Potentialkurven im zweidimensionalen EK- R-Koordinatensystem reduzieren, bilden.
der vier energetisch tiefsten Elektronenzustände j = 0, 1, 2, 3 berechnet wurden, die dann die Potentialflächen, die sich hier natürlich auf Potentialkurven im zweidimensionalen EK- R-Koordinatensystem reduzieren, bilden.
Die Masse der Atomkerne kommt in der Schrödinger-Gleichung (2-7) für die Elektronenzustandsfunktionen (2-5) nicht vor, d. h. das Ergebnis hängt nur von den Kernladungen und nicht von den Kernmassen ab: Gleiche Moleküle mit verschiedenen Isotopen haben jeweils dieselbe Energiehyperfläche.
Abb. 2-1. Die Potentialkurven
der vier energetisch tiefsten Elektronenzustände (j = 0, 1, 2, 3) des H2- Moleküls; diese Zustände können für kleinere Werte R näherungsweise durch die Konfigurationen 1σσ, 3σσ*, 1σσ* und 1σ*σ* beschrieben werden; bei größeren Werten R bis hin zur Dissoziation muß die Konfigurationswechselwirkung berücksichtigt werden (vgl. Kap. 2.3.1), wobei die beiden energetisch tieferen Zustände zu zwei H-Radikalen und die beiden energetisch höheren Zustände zum H+ und H− dissoziieren
Die Berechnung der Funktionen (2-5) der Elektronenzustände beruht also darauf, daß das Kerngerüst jeweils als ruhend angenommen wird, was dem Heisenbergschen Unschärfeprinzip und damit einem der wesentlichsten Prinzipien der Natur widerspricht. Quantenmechanisch müssen die Kernbewegungen, also die molekularen Schwingungen und die dazu korrespondierenden Aufenthaltswahrscheinlichkeiten der Kerne, durch die Schwingungsfunktion (2-6) beschrieben werden. Diese Schwingungsfunktion enthält nur die Kernkoordinaten, obwohl sich natürlich bei molekularen Schwingungen, also Änderungen der Kernkoordinaten, auch die Elektronenkoordinaten ändern. Da aber die Elektronen (im Rahmen der Näherung!) verzögerungsfrei den Kernbewegungen folgen, ist die Aufenthaltswahrscheinlichkeit der Elektronen zu jedem Moment den Kernbewegungen angeglichen und durch die Zustandsfunktion (2-5) des jeweiligen Elektronenzustandes j für die jeweilige augenblickliche Kernanordnung gegeben. Mit anderen Worten: Eine bestimmte Kernanordnung im Verlauf einer molekularen Schwingung bedingt augenblicklich die Anordnung und Energie der Elektronen, die durch die stationäre elektronische Zustandsfunktion (2-5) für diese Kernanordnung gemäß Kap.2.2.1.1 gegeben sind.
Die Funktion (2-6) die den stationären Zustand v der Kerne im Elektronenzustand j beschreibt, ist Lösung der zeitunabhängigen Schrödinger-Gleichung (2-8)
(2-8) 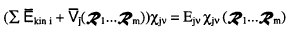
wobei der Operator der kinetischen Energie des Atomkernes i, und  der Operator der potentiellen Energie der Kerne ist. Diese potentielle Energie der Kerne ist durch die Potentialfläche als Lösung der Schrödinger-Gleichung(2-7) für den Elektronenzustand j gegeben; diese Energie ist - wie in 2.2.1.1 bereits erklärt - aus der Sicht der Kerne potentielle Energie, obwohl sie implizit auch die potentielle und kinetische Energie der Elektronen enthält4. Damit wird die Schrödinger-Gleichung(2-8) für die stationären Schwingungszustände (2-6) zu:
der Operator der potentiellen Energie der Kerne ist. Diese potentielle Energie der Kerne ist durch die Potentialfläche als Lösung der Schrödinger-Gleichung(2-7) für den Elektronenzustand j gegeben; diese Energie ist - wie in 2.2.1.1 bereits erklärt - aus der Sicht der Kerne potentielle Energie, obwohl sie implizit auch die potentielle und kinetische Energie der Elektronen enthält4. Damit wird die Schrödinger-Gleichung(2-8) für die stationären Schwingungszustände (2-6) zu:
(2-9) 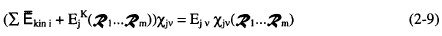
Deren Lösung liefert dann das bekannte Ergebnis, daß in einem Potentialtopf nicht alle Energiewerte der Energiehyperfläche erlaubt sind, sondern nur die diskreten stationären Energien EjV der erlaubten Kernschwingungen v = 0, 1, 2… des Elektronenzustandes j. Diese stationären Energien EjV enthalten nicht nur die kinetische und potentielle Energie der Kerne, für die die Schrödinger-Gleichung (2-9) explizit gelöst wird, sondern auch die kinetische und potentielle Energie der Elektronen, die ja implizit im Operator  der potentiellen Energie der Kerne in (2-9) enthalten ist. Damit ist die diskrete stationäre Energie Ejv die Gesamtenergie des Systems.
der potentiellen Energie der Kerne in (2-9) enthalten ist. Damit ist die diskrete stationäre Energie Ejv die Gesamtenergie des Systems.
Gelegentlich hört oder liest man, die Bedeutung der Born-Oppenheimer-Approximation liege darin, daß der rechnerische Aufwand bei der Lösung der Schrödinger-Gleichung reduziert werde, denn bei einem Molekül aus m Kernen und n Elektronen werde anstelle eines (m+n)-Teilchenproblems zuerst ein n-Teilchenproblem für die Funktionen (2-5) der stationären Elektronenzustände j, und dann ein m-Teilchenproblem für die stationären Schwingungszustände (2-6) gelöst. Diese Reduktion des rechnerischen Aufwandes mag früher wesentlich und wichtig gewesen sein, ist heute aber im Zeitalter leistungsfähiger Rechner kein gewichtiges Argument mehr für die Notwendigkeit der Born-Oppenheimer-Approximation. Wichtig ist vielmehr, daß die für die Diskussion und das Verständnis chemischer Probleme elementaren Begriffe Elektronenzustand, Potentialfläche und Schwingungszustand direkt aus der Born-Oppenheimer-Näherung folgen und ohne diese gar nicht definiert wären. Und ohne diese Begriffe wäre die konzeptionelle Behandlung der Spektroskopie und der chemischen Reaktion in der heute üblichen Art undenkbar!
Denn nur so ist eine chemische Reaktion als Wanderung auf einer Potentialfläche entlang eines Reaktionspfades von einem Minimum über einen Sattelpunkt (Übergangszustand) in ein anderes Minimum leicht vorstellbar und interpretierbar (vgl. Kap. 2.2.2). Und nur so lassen sich die verschiedenen, energetisch häufig dicht an dicht beieinander liegenden zahlreichen stationären Zustände eines Moleküls aufteilen in zum einen Elektronenzustände, die im Rahmen der Orbitalapproximation (vgl. Kap. 2.3) durch Elektronenkonfigurationen (bzw. deren Slater-Determinanten) repräsentiert werden, und zum anderen Schwingungszustände, die durch die Schwingungsquantenzahlen v definiert sind.
Die Geometrie eines H2-Moleküls ist durch die Angabe des Abstandes R der beiden Kerne eindeutig gegeben. Die Energien  als Lösungen der Schrödingergleichung (2-7) lassen sich als Funktion einer geometrischen Variablen auftragen, wie dies in Abb. 2-1 gezeigt wird; es handelt sich um eine zweidimensionale Darstellung.
als Lösungen der Schrödingergleichung (2-7) lassen sich als Funktion einer geometrischen Variablen auftragen, wie dies in Abb. 2-1 gezeigt wird; es handelt sich um eine zweidimensionale Darstellung.
Im Falle eines Systems mit zwei geometrischen Freiheitsgraden f1 und f2 läßt sich die Energie EK eines bestimmten elektronischen Zustandes, etwa des Grundzustandes, mittels isoenergetischer Linien graphisch dar- und damit auch sehr bildhaft in Form einer dreidimensionalen Potentialfläche vorstellen, entsprechend den Höhenlinien einer Landkarte mit der dazu korrespondierenden Landschaft mit Tälern, Bergen und Pässen.
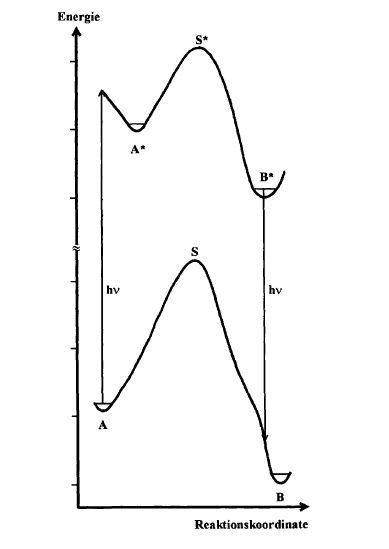
Abb. 2-2 zeigt derartige isoenergetische Linien für den elektronischen Grundzustand (unten) und den ersten angeregten Elektronenzustand (oben) eines fiktiven Systems, wobei die Schwingungsniveaus nicht eingezeichnet sind. Auf der Potentialfläche des Grundzustandes stellen die Punkte A und B Minima dar, also jeweils Kernanordnungen stabiler chemischer Verbindungen. Die thermische Reaktion A → B verläuft nach dem Prinzip des geringsten „Aufwandes“ entlang des gestrichelt gezeichneten Reaktionspfades über den Sattelpunkt S, der den Übergangszustand TS der Reaktion repräsentiert. Projiziert man den vertikalen Schnitt durch die Potentialfläche entlang des Reaktionspfades auf eine Ebene, dann wird die in Abb. 2-3 unten gezeigte Darstellung erhalten, wobei die Abszisse als Reaktionskoordinate bezeichnet wird; die Reaktionskoordinate subsummiert die gleichzeitige Änderung beider geometrischer Freiheitsgrade fi und f2. Nebst der thermischen Reaktion A → B ist aber auch die photochemische Umsetzung von A zu B denkbar: A wird durch Absorption eines nach Einstein energetisch „passenden“ Photons (vgl. Kap. 2.4) zum schwingungsangeregten Minimum A* auf der Potentialfläche des angeregten Elektronenzustandes angeregt. Von dort aus kann die Reaktion entlang des gestrichelt gezeichneten Reaktionspfades auf der Potentialfläche des angeregten Zustandes über den Sattelpunkt S* zum Minimum B* ablaufen, das - etwa unter Abgabe eines Photons - zum schwingungsangeregten B desakti viert5.
Abb. 2-2. Isoenergetische Linien der fiktiven dreidimen-sionalen Potentiallandschaft eines Grundzustandes (unten) und des dazu korrespondierenden elektronisch tiefsten angeregten Zustandes (oben) als Funktion zweier geometrischer Freiheitsgrade fi und f2. Die isoenergetischen Linien unter-scheiden sich jeweils um eine Energieeinheit; B bzw. B* sind jeweils absolute Minima, A und A* relative Minima, und C bzw. C* Maxima
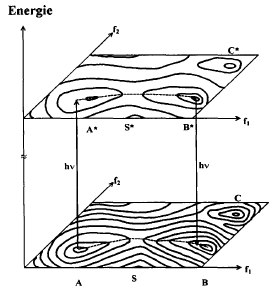
Die Projektion des senkrechten Schnittes durch die Potentialfläche des angeregten Zustandes entlang dieses Reaktionspfades auf eine Ebene ist in Abb. 2-3 oben gegen die Reaktionskoordinate aufgetragen.
Diese photochemische Reaktion wird allgemein durch die Reaktionsfomel A + hv → B beschrieben. Diese Formulierung sollte man aber nicht benützen, da intermediär noch weitere Minima, also mehr oder weniger stabile Strukturen auftreten, nämlich A* und B*, die nicht einfach unterschlagen werden sollten; besser, weil wirklichkeitsgetreuer, ist deshalb folgende Formulierung für diese photochemische Reaktion: A + hv → A* → B* → B + hv.
Minima auf einer Potentialfläche („Krater“) sind dadurch gekennzeichnet, daß jede beliebige Änderung der Geometrie eine Anhebung der Energie bewirkt, Maxima („Gipfel“) dadurch, daß jede Änderung der Geometrie eine Energieabsenkung bewirkt, und Sattelpunkte dadurch, daß sie bezüglich des Reaktionspfades Maxima, bezüglich aller anderer senkrechter Schnitte durch die Potentialfläche aber Minima darstellen.
Nun wird man sich fragen, warum ein fiktives, warum kein reales, wirklich vorstellbares und vorzeigbares Beispiel für die Abbildungen 2-2 und 2-3 benutzt wird. Nun, es gibt keines! Bereits im Falle eines dreiatomigen Moleküls hängt die potentielle Energie von drei Freiheitsgraden, und im Falle eines Moleküls mit m Atomkernen von (3m-6) Freiheitsgraden ab, wäre also die Potential „fläche“ im vierdimensionalen, bzw. im (3m-6+l)-dimensionalen zwar (tabellarisch) dar-, aber nicht mehr (bildlich) vorstellbar. Aber auch im (3m-6+l)-dimensionalen gibt es „Punkte“, bei denen jede beliebige Geometrieänderung die Energie ansteigen läßt (Minima auf der Potential„fläche“), gibt es zwischen diesen Minima Reaktions„pfade“, die über Sattel „punkte“ verlaufen, die wieder dadurch gekennzeichnet sind, daß ihre Energie bezüglich Geometrieänderungen entlang des Reaktionspfades maximal, bezüglich aller anderer Geometrieänderungen aber minimal ist. Und wieder kann der „senkrechte“ Schnitt durch die multidimensionale Potentialfläche entlang des Reaktionspfades auf eine zweidimensionale (Papier)-Ebene projiziert werden, kann also die Energie entlang des multidimensionalen Reaktionspfades gegen die Reaktionskoordinate aufgetragen werden. Obwohl die Reaktionskoordinate alle (also maximal 3m-6) Geometrieänderungen subsummiert, wird häufig einer der 3m-6 geometrischen Freiheitsgrade explizit an der Reaktionskoordinate angegeben, etwa einer, der sich besonders drastisch ändert, oder einer, der für den Verlauf der Reaktion besonders charakteristisch ist.
Abb. 2-3. Die zu Abb. 2-2 korrespondierenden Reaktionskoordinaten
eineeinein2ein2einen2