
Contents
Weitere Titel für Chromatographie-Einsteiger
Konrad Grob
Split and Splitless Injection for Quantitative Gas Chromatography
(inkl. CD-ROM)
2001
ISBN 3-527-29879-7
Bruno Kolb, L. S. Ettre
Static Headspace-Gas Chromatography
1998
ISBN: 0-471-19238-4 (John Wiley & Sons)
Michael Oehme
Praktische Einführung in GC/MS-Analytik mit Quadrupolen
1996
ISBN: 3-527-29718-9
Dean Rood
A Practical Guide to the Care, Maintenance and Troubleshooting of Capillary Gas Chromatographie Systems
1998
ISBN: 3-527-29750-2
Journal of Separation Science
ISSN: 1615-9306

Bruno Kolb
Im Weingärtle
D-88696 Owingen
1. Auflage 1999
2. Auflage 2002
1. Nachdruck 2005
2. Nachdruck 2006
Bibliografische Information Der Deutschen Bibliothek
Die Deutsche Bibliothek verzeichnet diese Publikation in der Deutschen Nationalbibliografie; detaillierte bibliografische Daten sind im Internet über <> abrufbar.
© 2003 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
ISBN 3-527-30687-0
Gedruckt auf säurefreiem Papier.
Alle Rechte, insbesondere die der Übersetzung in andere Sprachen, vorbehalten. Kein Teil dieses Buches darf ohne schriftliche Genehmigung des Verlages in irgendeiner Form – durch Photokopie, Mikroverfilmung oder irgendein anderes Verfahren – reproduziert oder in eine von Maschinen, insbesondere von Datenverarbeitungsmaschinen, verwendbare Sprache übertragen oder übersetzt werden. Die Wiedergabe von Warenbezeichnungen, Handelsnamen oder sonstigen Kennzeichen in diesem Buch berechtigt nicht zu der Annahme, daß diese von jedermann frei benutzt werden dürfen. Vielmehr kann es sich auch dann um eingetragene Warenzeichen oder sonstige gesetzlich geschützte Kennzeichen handeln, wenn sie nicht eigens als solche markiert sind.
All rights reserved (including those of translation in other languages). No part of this book may be reproduced in any form – by photoprinting, microfilm, or any other means – nor transmitted or translated into machine language without written permission from the publishers. Registered names, trademarks, etc. used in this book, even when not specifically marked as such, are not to be considered unprotected by law.
Print ISBN 9783527306879
Epdf ISBN 978-3-527-66296-8
Epub ISBN 978-3-527-66295-1
Mobi ISBN 978-3-527-66294-4
Vorwort zur 2. Auflage
Die vorliegende Einführung in die Gaschromatographie geht zurück auf eine Zusammenstellung von Vorlagen für Dias und Folien zu zahlreichen Kursen und Seminaren des Autors, einschließlich einer Vorlesungsreihe an der Universität Konstanz. Dieses Bildmaterial war für den Zweck als Kursunterlage zunächst nur mit kurzen Kommentaren versehen. Daher kommt der Titel dieses Buches: „Gaschromatographie in Bildern“. Das Konzept, jeweils eine Bildseite einer, nun aber erweiterten, Textseite gegenüberzustellen, hat eine so freundliche Aufnahme gefunden, dass eine 2. Auflage erforderlich wurde. Darin sind Anregungen berücksichtigt, die der Autor von verschiedenen Seiten erhalten hat und für die er dankbar ist. Eine wesentliche Ergänzung ist das neu hinzugekommene Kapitel über die Verwendung von Massenspektrometehi als GC-Detektoren. In der 1. Ausgabe wurde die GC/MS-Kopplung absichtlich weggelassen, weil dieses Buch in der Schriftenreihe Handbibliothek Chemie zusammen mit dem Buch von M. Oehme („Praktische Einführung in die GC/MS-Analytik mit Quadrupolen“) erscheinen sollte. Auch schien es dem Autor nicht möglich, das Kapitel GC/MS im vorgegebenen Umfang erschöpfend zu behandeln. Diese Bedenken bestehen zwar nach wie vor und sind der Grund, warum sich der Autor auf die beiden Typen von Massenspektrometern beschränkt hat, die am häufigsten in Routine-Labors vertreten sind, nämlich den Quadrupol- und den Ion-Trap Geräten. Ebenfalls wurde darauf verzichtet, die Kopplung von Infrarotspektrometern mit der GC (GC/FTIR) hier aufzunehmen.
Auch bei dieser überarbeiteten Auflage ist nicht auszuschließen, dass sich Fehler oder missverständliche Formulierungen eingeschlichen haben; für entsprechende Hinweise von aufmerksamen Lesern ist der Autor dankbar.
Oktober 2002
Bruno Kolb
Vorwort zur 1. Auflage
Gaschromatographie (GC) kann man als Wissenschaft, aber auch als praktisches Laborhandwerk betreiben. Als Wissenschaft war sie außerordentlich nützlich und erfolgreich, da es mit ihrer Hilfe erstmals gelang, eine allgemeine Theorie der Chromatographie zu konzipieren, die auf der Chemie der zwischenmolekularen Wechselwirkungen, der Physik der Transport- und der Kinetik der Austauschvorgänge basiert. Andererseits ist die GC auch leicht verständlich und der Grund dafür ist ziemlich einfach: Trennungen werden durch die Flüchtigkeit der Stoffe und durch ihre Polarität hervorgerufen. Dampfdrücke, d. h. praktisch Siedepunkte, sowie Stoffpolaritäten sind Begriffe, die dem Chemiker vertraut sind und an denen er sich orientieren kann. Der praktische Anwender muß sich daher nicht notwendigerweise mit der Theorie befassen, um erfolgreich damit arbeiten zu können. Die praktischen Folgerungen aus der Theorie haben die Hersteller von Geräten, Säulen und Zubehören gezogen und dieses Know-how wird vom Käufer – meist unbewußt – erworben. Man kann daher GC auch ohne jedes Verständnis für die chromatographischen Grundlagen betreiben, wenn man lediglich Standardvorschriften (z. B. SOPs: „standard operating procedures“) abarbeitet, in denen jedes Detail und jeder Handgriff penibel vorgeschrieben sind. Wozu also ein einführendes Lehrbuch? Die Antwort ergibt sich aus der Erfahrung des Autors aus zahlreichen Einführungskursen zur GC, wonach das Abarbeiten von SOPs ohne Verständnis für die vorgeschriebenen Maßnahmen meist als unbefriedigend empfunden wird.
Diese Einführung richtet sich daher an alle Praktiker der verschiedensten Disziplinen, die sich nicht ausschließlich mit der GC beschäftigen können, die sich aber dennoch über die wesentlichen Grundlagen und Techniken informieren möchten, ohne sich in feinere Details zu verlieren. Aus diesem Grund wurde die Darstellung mit möglichst vielen Bildern gewählt. Auch bei der Beschreibung von chemischen Wechselwirkungen mit stationären Phasen oder von Reaktionen in Detektoren konnten nur allgemein akzeptierte Anschauungen berücksichtigt werden, die für das Verständnis der beschriebenen Vorgänge ausreichen, ohne jedoch auf manchmal alternativ diskutierte Ansichten einzugehen.
Zum Schluß möchte ich mich herzlich bei Herrn Ui Servos (Perkin-Elmer-Büro Düsseldorf) für zahlreiche praktische Empfehlungen bedanken. Dank gebührt auch Herrn PD Dr. Wolfgang Dünges für die Anregung, dieses Buch zu schreiben.
April 1999
Bruno Kolb
Die Nomenklatur der Gaschromatographie (GC) ist besonders in der deutschsprachigen Literatur – historisch bedingt – nicht einheitlich. Vom Arbeitskreis Chromatographie der Fachgruppe Analytische Chemie in der Gesellschaft Deutscher Chemiker (bearbeitet von H. Engelhardt und L. Rohrschneider) wurden entsprechend der IUPAC-Empfehlung „Nomenclature for Chromatography“ (Pure & Appl Chem. Vol. 65, 819–872 (1993)) Vorschläge für eine analoge deutschsprachige Nomenklatur herausgegeben. Die in der vorliegenden Schrift verwendeten Begriffe und Symbole folgen mit wenigen geringfügigen Ausnahmen diesen Empfehlungen.
Schwierigkeiten hat es immer mit einer zutreffenden Übersetzung der Bezeichnung „open tubular columns“ gegeben. Zeitweilig wurde dafür die wörtliche Übersetzung offene Röhren verwendet, dann offene Kapillarsäulen. Auch die Bezeichnung Kapillar-Trennsäule findet sich. Vom Autor dieser Schrift wird an sich die von R. Kaiser empfohlene prägnante Bezeichnung Trennkapillare sehr geschätzt, aber zugunsten der in den o. g. Empfehlungen benutzten Bezeichnung Kapillarsäule aufgegeben. Für die weitere Unterteilung in Film- bzw. Schichtkapillarsäulen wird in dieser Schrift aber meist die kürzere Form Film- bzw. Schichtkapillare verwendet, da aus diesen Bezeichnungen eindeutig hervorgeht, daß sie eine stationäre Phase enthalten, da sie andernfalls nicht trennen könnten.
In den Anfängen der GC wurde versucht, tabellierbare und normierte Retentionsdaten zu bestimmen, wozu alle apparativen und experimentellen Einflußgrößen auf die Retention eines Peaks herausgerechnet wurden. Durch Multiplikation der Retentionszeit mit der Flußgeschwindigkeit des Trägergases wurden zunächst entsprechende Retentionsvolumina bestimmt, auf die Menge stationärer Phase normiert und auf die Standardtemperatur 0 °C bezogen (spezifisches Retentionsvolumen Vg). Die Erwartungen, damit eine Identifizierung aufgrund tabellierter Datensammlungen zu ermöglichen, haben sich allerdings nicht erfüllt. Infolgedessen wird hier der Begriff des Retentionsvolumens nicht weiter verwendet. Er spielt auch bei der praktischen Anwendung der Gaschromatographie keine Rolle. Durchgesetzt haben sich dagegen relative Retentionsangaben (relative Retention, Retentionsindex); dafür genügen aber allein die Retentionszeiten, wie sie vom Integrator bzw. dem Datensystem gemessen werden.
| chromatographische Systeme | mobile Phase |
| Flüssigchromatographie: | Flüssigkeit |
| Gaschromatographie: | Gas (Trägergas) |
| Bezeichnung | stationäre Phase |
| Gas-Flüssig-Chromatographie (Verteilungs-GC): | Flüssigkeit |
| Gas-Fest-Chromatographie (Adsorptions-GC): | Feststoff |
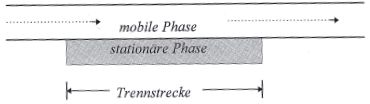
Chromatographie ist ein physikalisch-chemisches Trennverfahren, bei dem sich die zu trennenden Substanzen (Analyten) zwischen zwei nicht mischbaren Phasen verteilen. Der Begriff Phase bezeichnet einen stofflichen Aggregatzustand und eine chromatographische Phase kann daher ein Feststoff, eine Flüssigkeit oder ein Gas sein. Eine der beiden Phasen ist stationär, während die andere die chromatographische Trennstrecke in einer Richtung durchströmt und als mobile Phase den Stofftransport bewirkt. Als mobile Phasen können nur fluide Stoffe eingesetzt werden, und durch sinnvolle Kombinationen ergibt sich daraus eine formale Einteilung in die Verfahren der Flüssigchromatographie und der Gaschromatographie.
Trennstrecke in der Gaschromatographie ist immer ein Rohr, das aus historischen Gründen als Säule bezeichnet wird. Bei einer gepackten Säule ist dieses Rohr mit einem feinkörnigen Füllmaterial dicht gestopft. Bei Kapillarsäulen ist dieses Rohr wesentlich länger und dünner und die stationäre Phase haftet als dünner Film oder als Schicht an der Wand der Kapillare selbst. Kapillarsäulen weisen daher einen offenen Längskanal auf („open-tubular columns“). Für die mobile Phase, das sog. Trägergas, werden Inertgase, wie z. B. Stickstoff, Helium, Argon oder Wasserstoff verwendet.
Bei der Gaschromatographie ergibt sich eine formale Klassifizierung nach den stofflichen Eigenschaften der stationären Phase. Diese kann ein festes Adsorbens sein (Adsorptions-Gaschromatographie), an dessen aktiver Oberfläche die flüchtigen Analyten durch reversible Adsorption festgehalten werden, oder sie besteht aus einer nichtflüchtigen Flüssigkeit, die als dünner Film auf der Oberfläche eines Trägers haftet oder an ihr chemisch gebunden ist. Die flüssige stationäre Phase wirkt als Lösemittel, in dem sich die flüchtigen Analyten teilweise lösen (Verteilungs-Gaschromatographie). Der Träger ist bei gepackten Säulen ein körniges und poröses Material (z. B. Kieselgur), im Fall von Kapillarsäulen die Wand der Kapillare selbst.
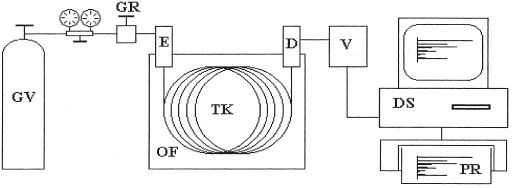
| GV | Gasversorgung durch Druckgasflasche |
| GR | Gasregelung: Druckregler, Strömungsregler |
| E | Einlaßteil: Injektor, Gasprobengeber etc. |
| TK | Kapillarsäule oder gepackte Säule |
| OF | Ofen |
| D | Detektor |
| V | Verstärker |
| DS | Datensystem |
| PR | Printer/Plotter |
Das Trägergas wird einem Druckbehälter über Reduzier- und Regelventile entnommen und druck- oder strömungsgeregelt dem Einlaßteil eines Gaschromatographen zugeführt. Das kann je nach Art der zu analysierenden Probe ein Injektor für flüssige Proben sein oder eine Gasdosierschleife (s. Abschnitt 7.11); auch können über Adapter zahlreiche Zubehöre (Pyrolysator, Thermodesorber, Headspacesampler, „purge-and-trap“- Vorrichtung, etc.) angeschlossen werden. Die Trennsäule befindet sich in einem thermostatisierten Ofen. Ihr Ende ist mit dem Detektor verbunden, der die Änderung in der Zusammensetzung des Trägergases beim Erscheinen einer getrennten Substanz zuerst registriert, dann in ein elektrisches Signal umwandelt und über einen Verstärker dem Datensystem (Computer, Integrator) zur weiteren Auswertung übermittelt.
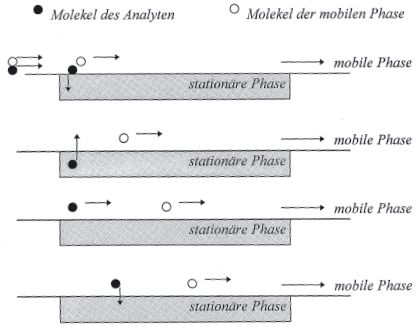
Ursache der chromatographischen Trennung ist die Retention: Die Stoffmoleküle werden vom Trägergas durch die Trennstrecke transportiert. Betrachten wir jeweils ein Molekel (Atom oder Molekül) der mobilen Gasphase und des flüchtigen Analyten, die beide zur gleichen Zeit in die Säule eintreten sollen. Sie unterscheiden sich in ihrem Verhalten insofern, als daß das Molekel der mobilen Phase stetig durch die Säule wandert, während das Molekel des Analyten sich dem Transport zeitweise entzieht, indem es in die stationäre Phase hinein und wieder zurückdiffundiert oder von deren Oberfläche kurzzeitig durch Adsorption festgehalten wird. Infolgedessen wird die Trennstrecke vom Molekel des Analyten insgesamt langsamer zurückgelegt als von der mobilen Phase. Diese Verzögerung der Wanderungsgeschwindigkeit wird als chromatographische Retention bezeichnet. Tatsächlich aber wandert das Molekel des Analyten immer mit der Geschwindigkeit des Trägergases, solange es sich darin aufhält. Nur für das Kollektiv der Molekel ergibt sich insgesamt eine langsamere Wanderungsgeschwindigkeit.
(1.1) 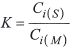
(1.2) 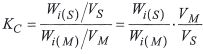
(1.3) 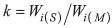
(1.4) 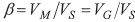
| Ci(S) | Konzentration des Analyten i in der stationären Phase |
| Ci(M) | Konzentration des Analyten i in der mobilen Gasphase |
| Wi(S) | Menge des Analyten i in der stationären Phase |
| Wi(M) | Menge des Analyten i in der mobilen Gasphase |
| VS | Volumen der stationären Phase |
| VM | Volumen der mobilen Gasphase (=VG) |
| k | Mengenverhältnis |
| ß | Phasenverhältnis |
Die Aufteilung eines flüchtigen Analyten zwischen den beiden Phasen – der Verteilungsvorgang – kommt dadurch zustande, daß die Molekel ständig zwischen den beiden Phasen hin und her pendeln. Würden wir nun den Fluß des Trägergases abstellen, würde sich nach einiger Zeit aus dieser beidseitigen Diffusion ein Gleichgewicht einstellen, das durch eine Gleichgewichtskonstante beschrieben werden kann. Wenn wir daraufhin das Trägergas wieder fließen lassen, werden die Molekel in der Gasphase von der Strömung erfaßt und mitgenommen, die Querdiffusion jedoch hält beide Molekelhaufen zusammen: An der Rückfront des Molekelhaufens ist nur noch der Übergang von der stationären Phase in die Gasphase möglich. Umgekehrt besteht an der Front für die Molekel nur die Möglichkeit, aus der Gasphase in die stationäre Phase hineinzudiffundieren; es sind noch keine Molekel da, die rückdiffimdieren könnten. Nur in dem Bereich, in dem sich beide Molekelhaufen überlappen, liegt der Gleichgewichtszustand vor. Auf diese Weise wälzt sich sozusagen die Molekelzone durch die Säule, angetrieben durch den Fluß des Trägergases, aber langsamer als dieses. Die verzögerte chromatographische Wanderung wird im Grunde genommen durch die permanente Störung des Gleichgewichts hervorgerufen. Das Kollektiv tritt dann am Ende der Säule als Bande in den Detektor, wo es als sog. Peak registriert und vermessen wird.
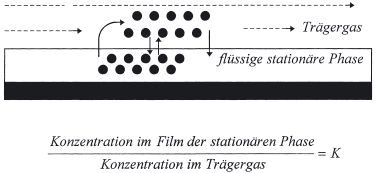
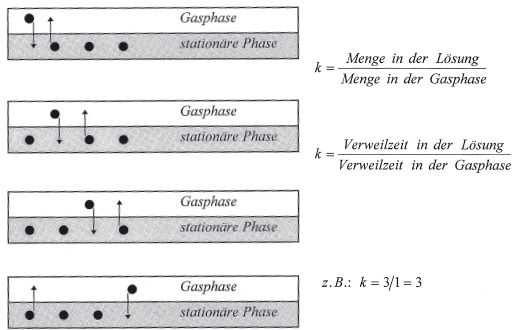
Die Gleichgewichtskonstante wird als Verteilungskonstante (K), häufig auch als Verteilungskoeffizient, bezeichnet: Sie gibt an, um wieviel größer die Konzentration eines Stoffs in der stationären Phase (CS) gegenüber der in der mobilen Gasphase (CM) ist. Im Fall der Gas-Flüssig-Chromatographie werden die Konzentrationen als Menge pro Volumen der jeweiligen Phase angegeben. Bei der Gaschromatographie ist das Volumen der mobilen Phase (VM) in der Trennsäule identisch mit dem Volumen der Gasphase (VG). Daher setzt sich hier die mit Kc bezeichnete Verteilungskonstante zusammen aus dem Mengenverhältnis (k) des Analyten (i) in den beiden Phase und deren Volumenverhältnis (VG/Vs), das auch als Phasenverhältnis (ß) bezeichnet wird. Es ist eine wichtige Säulenkonstante.
Das Mengenverhältnis (k) wird auch als Retentionsfaktor, Massenverteilungsfaktor oder Kapazitätsfaktor bezeichnet. Diese vielfaltigen Bezeichnungen entsprechen der zentralen Bedeutung dieser Größe in der GC.
Besteht dagegen die stationäre Phase aus einem Feststoff, kann statt auf das Volumen auch auf das Gewicht oder die spezifische Oberfläche Bezug genommen werden. Dieser Fall wird in Kapitel 4 behandelt.
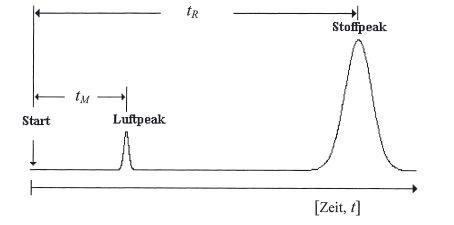
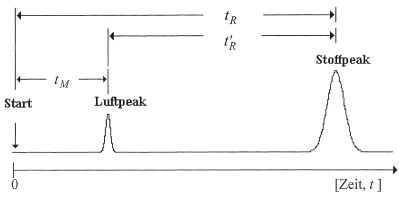
| tR | = gesamte Verweilzeit eines Stoffs in der Säule = Gesamtretentionszeit |
| tM | = Durchflußzeit des Luftpeaks = Verweilzeit aller Stoffe in der Gasphase |
Zwei Stoffe werden getrennt, wenn ihre Verteilungskonstanten verschieden sind. Da das Phasenverhältnis eine Apparatekonstante der jeweiligen Säule und daher für alle Stoffe gleich ist, können wir die weitere Diskussion mit dem Mengenverhältnis (k) führen. Ist z. B. k gleich drei, dann ist die Menge dieses Stoffs in der stationären Phase dreimal so groß wie in der Gasphase. Auf jedes Molekel in der Gasphase treffen daher drei Molekel in der stationären Phase. Um das Gleichgewicht zu wahren, muß für jedes Molekel, das aus der Gasphase in die stationäre Phase diffundiert, ein anderes aus der stationären Phase in die Gasphase übertreten. Daraus folgt, daß in der stationären Phase jedes Molekel dreimal so lange warten muß, bis es wieder an die Reihe kommt. Infolgedessen beschreibt das Mengenverhältnis (k) auch das Verhältnis der Verweilzeiten eines Stoffs in den beiden Phasen. Das ist ein wichtiger Zusammenhang zwischen dem statischen thermodynamischen Gleichgewicht und der dynamischen Wanderung im chromatographischen System, da diese Verweilzeiten auch das Retentionsverhalten eines Stoffs bestimmen.
Die gesamte Verweildauer eines Stoffs in der Säule entspricht der Zeit von der spontanen Probenaufgabe bis zum Austreten des Peakmaximums aus der Säule und ist gleich der gesamten Retentionszeit (tR). Schwieriger wird es schon, diese Gesamtretentionszeit nun in die Verweilzeiten des Stoffs in der stationären und in der Gasphase aufzuschlüsseln.
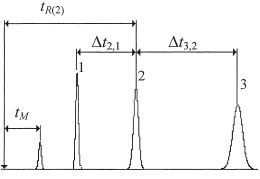
Die Verweilzeit in der Gasphase ist für alle Stoffe gleich und läßt sich mit Hilfe eines Stoffs bestimmen, der sich in der stationären Phase nicht löst und infolgedessen mit der Geschwindigkeit des Trägergases durch die Säule wandert. So ein Stoff ist z. B. ein Inertgas, wie Luft, wenn man eine Säule mit einer flüssigen stationären Phase benutzt. Man benötigt dann allerdings einen Detektor, der den Luftpeak (Inertpeak) mit der Durchflußzeit (tM), häufig auch als Totzeit bezeichnet, anzeigt. Alternativ wird im Fall eines Flammenionisationsdetektors (FID) Methan verwendet, unter der Annahme, daß es von der stationären Phase unter den gegebenen Bedingungen – höhere Temperatur, kurze Säule, dünner Film – nicht zurückgehalten wird und sich daher wie ein Inertstoff verhält. Im System des Retentionsindex kann die Durchflußzeit auch aus den Retentionszeiten von drei aufeinanderfolgenden n-Alkanen nach (s. S. 16) berechnet werden.
(1.5) 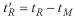
(1.6) 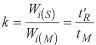
(1.7) 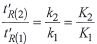
(1.8) 
(1.9) 
Die Verweilzeit eines Stoffs in der flüssigen stationären Phase kann nicht unmittelbar bestimmt werden. Sie errechnet sich als Differenz zwischen der Gesamtretentionszeit und der Durchflußzeit und wird als reduzierte Retentionszeit (t'R) bezeichnet.
Diese Berechnung ist insofern möglich, als die Verweilzeit in der Gasphase für alle Stoffe gleich ist. Befindet sich ein Stoff in der Gasphase, wandert er immer mit der Trägergasgeschwindigkeit weiter, und diese Wanderung wird nur durch die Verweilzeit in der stationären Phase unterbrochen. Für ein einzelnes Molekel gilt, daß es entweder mit der Geschwindigkeit des Trägergases transportiert wird oder überhaupt nicht, solange es sich nämlich in der stationären Phase aufhält.
Aus den Retentionszeiten lassen sich durch Multiplikation mit der Flußgeschwindigkeit des Trägergases auch die analogen Retentionsvolumina (Gesamtretentionsvolumen (VR), reduziertes Retentionsvolumen (V'R) und das Durchflußvolumen (VM)) angeben. Für die Peakerkennung werden aber sowohl zur qualitativen Identifizierung als auch für die quantitative Analyse immer die vom Datensystem bestimmten Retentionszeiten zugrundegelegt, daher verwenden wir hier ausschließlich diese Größen.
Da die Verweilzeit in der Gasphase für alle Stoffe dieselbe ist, sind zwei Stoffe chromatographisch nur trennbar, wenn sie sich unterschiedlich lange in der stationären Phase aufhalten und dadurch unterschiedlich lange Retentionszeiten aufweisen. Ihre Trennbarkeit läßt sich daher durch das Verhältnis der reduzierten Retentionszeiten beschreiben. Dieses Verhältnis wird als relative Retention (r) bezeichnet; es gibt gleichzeitig das Verhältnis aller davon abgeleiteten Größen wieder, wie z. B. das der Mengenverhältnisse (k) und der Verteilungskonstanten (K). Die relative Retention (r) wird bestimmt, indem man einen bekannten Stoff als Referenzsubstanz benutzt. Je nach Reihenfolge der Retention kann r daher größer oder kleiner als 1 sein.
Das gleiche Retentionsverhältnis, aber jetzt von zwei benachbarten Peaks im Chromatogramm wird auch als Trennfaktor (α) bezeichnet, wobei man definitionsgemäß den ersten Peak als Referenzpeak benutzt, wodurch α immer ≥ 1 wird. Der Trennfaktor (α) wird zur Beschreibung der Auflösung nach (s. S. 32) und damit zur Charakterisierung des Trennvermögens einer Säule verwendet.
(1.10) 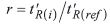
Eine Identifizierung getrennter Komponenten erfolgt im Prinzip immer mit entsprechenden Vergleichsstoffen unter den gleichen apparativen Bedingungen. Dazu kann man vor oder nach der Trennung einer Probe diese Vergleichsstoffe nacheinander chromatographieren und die Retentionszeiten direkt vergleichen (die Chromatogramme „übereinanderlegen“). Mit zunehmender Zahl von Stoffen wird der Aufwand unpraktikabel. Man kann das Verfahren abkürzen, wenn man nur einen Referenzstoff verwendet und diesen entweder separat unter den gleichen Bedingungen chromatographiert oder zur Probe zumischt. Die Retentionszeiten der Probenkomponenten werden dann auf diesen Referenzstoff bezogen und relativ dazu entweder als relative Retention oder als Retentionsindex angegeben. Bei letzterem werden mindestens zwei solcher Referenzstoffe benötigt.
Bei der relativen Retention (r) wird das Verhältnis der reduzierten Retentionszeiten des Analyten und des Referenzstoffs gebildet (s. ). Die apparativen Parameter, wie Strömungsgeschwindigkeit des Trägergases und Länge der Säule, sind in der Durchflußzeit (tM) enthalten und deren Einfluß auf die Retention ist damit eliminiert. Das Verhältnis der reduzierten Retentionszeiten des Analyten (t'R(i)) zum Referenzstoff (t'R(ref)) hängt dann nur noch vom Verhältnis der Verweilzeiten der beiden Stoffe in der stationären Phase ab. Es wird von der Temperatur und der Art der stationären Phase beeinflußt, die infolgedessen zusammen mit der relativen Retention (r) ebenfalls mitangegeben werden müssen. Die relative Retention ist daher – im Prinzip – unabhängig von apparativen Bedingungen (z. B. von unterschiedlichen Gasströmungen, unterschiedlich langen Säulen mit unterschiedlichen Filmdicken), und sie gilt gleichermaßen für Kapillarsäulen wie für gepackte Säulen. Damit sind die Retentionsdaten, z. B. aus verschiedenen Labors mit unterschiedlichen Geräten „vergleichbar“ und können daher für Identifizierungszwecke auch aus Tabellen entnommen werden. Verständlicherweise sind die an eigenen Geräten erstellten Retentionsdaten besser reproduzierbar als solche aus anderen Labors oder aus Literaturtabellen.
Der Trennfaktor (α) ist nur isotherm anwendbar und das gilt streng auch für die relative Retention (r). Letztere kann jedoch bei temperaturprogrammierter Arbeitsweise für Identifizierungszwecke mit entsprechenden Einschränkungen auch unter den folgenden Bedingungen benutzt werden:
(1.11) 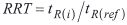
Wenn man nämlich für quantitative Analysen sowieso unter den gleichen apparativen Bedingungen einen Kalibrierstandard chromatographieren muß, der alle interessierenden Stoffe enthält, sind die Komponenten ja bekannt und die Zuordnung kann entweder durch unmittelbaren Vergleich der Gesamtretentionszeiten (tR) oder ebenfalls aufgrund von relativen Retentionsangaben erfolgen. Im letzteren Fall ist es aber dann zulässig und ausreichend, die Gesamtretentionszeiten (tR) zu verwenden, was mit Integratoren und Rechnern einfacher ist. Diese relative Angabe erfolgt durch die sog. relative Retentionszeit (RRT) („relative retention time“). Da für diese Aufgabenstellung nur die Reproduzierbarkeit der Retentionszeiten bei gleichbleibenden apparativen Bedingungen maßgebend ist, kann die Bestimmung auch bei temperaturprogrammierter Arbeitsweise erfolgen. Es geht damit aber die „Vergleichbarkeit“ verloren, d. h. diese Werte sind nicht auf Tabellenwerte anwendbar oder auf Messungen unter anderen instrumentellen Bedingungen übertragbar.
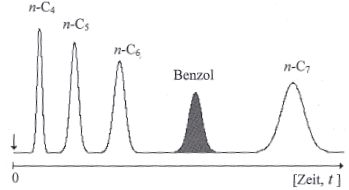
Bei Vielkomponentengemischen ist es sicherer, mehrere Referenzstoffe für die relativen Retentionsangaben zu verwenden. Auf dieser Vorstellung basiert der Retentionsindex (Kovats-Index). Das Bezugssystem ist hier die homologe Reihe der n-Alkane und damit wird der gesamte Temperaturbereich der GC erfaßt. Der interessierende Peak, z. B. von Benzol (B) wird von den beiden Referenzpeaks n-Hexan (n-C6) und n-Heptan (n-C7) flankiert. Den n-Alkanen werden unabhängig von der Art der stationären Phase und der Temperatur feste Indexwerte (I) zugeordnet, die sich aus der mit 100 multiplizierten Kohlenstoffzahl des jeweiligen n-Alkans ergeben:
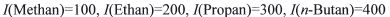
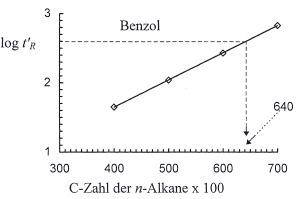
Das System beruht auf einem logarithmischen Maßstab, da die Retentionszeiten von Homologen bei isothermer Arbeitsweise logarithmisch zunehmen. Es besteht daher ein linearer Zusammenhang zwischen den Logarithmen der reduzierten Retentionszeiten der n-Alkane und ihrer C-Zahl, wie die graphische Darstellung zur Bestimmung des Retentionsindex von Benzol (I = 640) auf einer Squalan-Säule bei 60 °C zeigt.
(1.12) 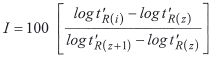
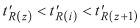
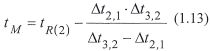
(1.14) 
(1.15) 
Die Retentionsindices werden nach nebenstehender Formel 1.12 aus den Logarithmen der reduzierten Retentionszeiten berechnet. Jede Komponente i mit ihrer reduzierten Retentionszeit (t'R(i)) liegt zwischen den Peaks der beiden benachbarten n-Alkane, wobei z die Zahl der C-Atome des n-Alkans ist, das vor der Komponente i eluiert und (z+1) die C- Zahl des entsprechenden n-Alkans, das danach kommt. Die für die Berechnung der reduzierten Retentionszeiten erforderliche Durchflußzeit (tM) kann im System des Retentionsindex, z. B. nach Peterson und Hirsch, (s. ) aus drei aufeinanderfolgenden n-Alkanen (1,2 und 3) berechnet werden.
Die Abhängigkeit der relativen Retention von der Temperatur ist logarithmisch und reziprok (s. ). Dagegen kann der Retentionsindex in einem nicht zu weiten Temperaturintervall nach linear extrapoliert werden. Die Angabe erfolgt in δI//10 °C und lautet z. B. für Benzol auf Squalan:

Der Retentionsindex bietet für die Zuordnung bei der quantitativen Analyse gegenüber der relativen Retention keinen Vorteil, er wird aber wegen seiner guten „Vergleichbarkeit“ zur Identifizierung von Proben unbekannter Zusammensetzung mittels tabellierter Daten bevorzugt. Allerdings ist durch die GC/MS-Kopplung die Bedeutung zur Stoffidentifizierung stark zurückgegangen, ebenso wie die Möglichkeit, Retentionsdaten von gaschromato- graphisch bisher nicht erfaßten Stoffen durch Addition von Indexincrementen zu berechnen. Der Retentionsindex wird meist nur noch in solchen Fällen eingesetzt, in denen – wie bei komplexen Kohlenwasserstoffgemischen – die Massenspektrometrie (MS) manchmal keine eindeutige Zuordnung der zahlreichen Isomeren zuläßt. Auch für Screeningzwecke eignet sich das Verfahren. Ein Beispiel dafür ist die Sammlung der Indices von > 1500 toxischen Stoffen auf einer unpolaren Säule für schnelles klinisches Screening in Vergiftungsfällen. Für diese Anwendung ist ein schneller Ausschluß von > 99 % der möglichen Giftstoffe besonders nützlich, um die wenigen noch verbleibenden Stoffe um so schneller zielgerichtet identifizieren zu können. Bedeutung hat das Index-System aber nach wie vor zur Charakterisierung von Säulenpolaritäten (s. Kapitel 3.3) durch das Rohrschneider/McReynolds-System.
Diese geläufige Bezeichnung wird hier beibehalten, obwohl relative Angaben prinzipiell dimensionslos sind.
M. L. Peterson and J. Hirsch, J.Lipid Res. 1 (1959) 132.
Gas-Chromatographic Retention Indices of Toxicologically Relevant Substances on SE-30 or OV-1; DFG und TIAFT (Hrsg.), VCH Verlagsgesellschaft mbH, Weinheim, 1985.
Die Bezeichnung Säule für eine gaschromatographische Trennstrecke ist historisch bedingt: sie stammt aus der frühen Zeit der Flüssigkeitschromatographie, als körnige Adsorbentien in kurze und dicke Röhren gestopft wurden, die durchaus Ähnlichkeit mit stämmigen Säulen hatten. Auch die Gaschromatographie hat mit derartigen Säulen, den sog. gepackten Säulen begonnen. Diese sind nach wie vor für bewährte Anwendungen, besonders für routinemäßige Gasanalysen, wegen ihrer Robustheit und Langzeitstabilität beliebt. Ihr Durchmesser beträgt üblicherweise 1/4" (6.35 mm) oder 1/8" (3.18 mm) und bei den sog. mikrogepackten Säulen < 1 mm. Das Trennvermögen dieser Säulen ist jedoch begrenzt und die Forderung nach höherer Auflösung wurde mit der Einführung der Kapillarsäulen durch M. Golay (1957) erfüllt. Bedingt durch den offenen Längskanal („open tubular column“) und den damit verbundenen geringen Druckabfall war es möglich, wesentlich längere Säulen zu verwenden. Trotzdem sind die Analysenzeiten wegen der geringen Menge an stationärer Phase in den dünnen Kapillaren akzeptabel, die aber andererseits eine sehr geringe Probenkapazität und eine damit verbundene geringere Nachweisempfindlichkeit zur Folge hat. Die kleine Probenkapazität zwang dazu, einen Probenteiler („splitter“) zu benutzen, mit dem eine noch handhabbare Probenmenge (μL-Volumina) vor der Säule soweit geteilt wird, daß nur wenige Prozent der verdampften Probenmenge tatsächlich in die Säule gelangen. Die damit verbundene geringe Nachweisempfindlichkeit, ein gravierender Nachteil besonders für Spurenanalysen, wurde durch die sog. splitlose Probenaufgabe durch K. Grob entscheidend verbessert. Im Verlauf der weiteren Entwicklung wurden dann verschiedenen Techniken zur kalten Probenaufgabe eingeführt, die sowohl mit als auch ohne Split betrieben werden. Für den konkreten praktischen Fall ist eine Auswahl aus diesen zahlreichen Möglichkeiten nicht weniger schwierig als die Wahl der richtigen Säule. Der eigentliche Durchbruch für die Kapillarsäulen erfolgte jedoch erst durch die Fused-Silica-Kapillaren, die 1975 von R. Dandeneu und E.H. Zerenner eingeführt wurden und wegen ihrer Flexibilität die zerbrechlichen Glaskapillaren und wegen ihrer Inertheit und ihrer geringen Masse die Stahloder Kupferkapillaren ersetzten.
| mesh | μm-Bereich |
| 60/80 | 250–177 |
| 80/100 | 177–149 |
| 100/120 | 149–125 |
Bei gepackten Säulen ist ein Rohr aus Metall, Glas oder Kunststoff (z. B. PTFE) mit dem sog. Säulenfüllmaterial dicht gefüllt, d. h. gepackt. Die äußere Gestalt richtet sich nach der Form des Ofenraums des jeweiligen Gaschromatographen.
Im Fall der Adsorptions-Gaschromatographie besteht das Füllmaterial aus dem feinkörnigen Adsorbens. Im Fall der Verteilungs-Gaschromatographie besteht es aus einem porösen Trägermaterial, dessen Oberfläche mit dem Film der flüssigen stationären Phase imprägniert ist. Meist wird dazu Diatomeenerde verwendet; das ist ein Gemenge aus den porösen Gerüststrukturen und Schalen von Kieselalgen (Radiolaren), das zu etwa 90 % aus reinem SiO2