Methods and Principles in Medicinal Chemistry
Edited by R. Mannhold, H. Kubinyi, G. Folkers
Editorial Board
H. Buschmann, H. Timmerman, H. van de Waterbeemd, T. Wieland
Previous Volumes of this Series:
Gohlke, Holger (Ed.)
Protein-Ligand Interactions
2012
ISBN: 978-3-527-32966-3
Vol. 53
Kappe, C. Oliver / Stadler, Alexander / Dallinger, Doris
Microwaves in Organic and Medicinal Chemistry
Second, Completely Revised and Enlarged Edition
2012
ISBN: 978-3-527-33185-7
Vol. 52
Smith, Dennis A. / Allerton, Charlotte / Kalgutkar, Amit S. / van de Waterbeemd, Han / Walker, Don K.
Pharmacokinetics and Metabolism in Drug Design
Third, Revised and Updated Edition
2012
ISBN: 978-3-527-32954-0
Vol. 51
De Clercq, Erik (Ed.)
Antiviral Drug Strategies
2011
ISBN: 978-3-527-32696-9
Vol. 50
Klebl, Bert / Müller, Gerhard / Hamacher, Michael (Eds.)
Protein Kinases as Drug Targets
2011
ISBN: 978-3-527-31790-5
Vol. 49
Sotriffer, Christoph (Ed.)
Virtual Screening
Principles, Challenges, and Practical Guidelines
2011
ISBN: 978-3-527-32636-5
Vol. 48
Rautio, Jarkko (Ed.)
Prodrugs and Targeted Delivery
Towards Better ADME Properties
2011
ISBN: 978-3-527-32603-7
Vol. 47
Smit, Martine J. / Lira, Sergio A. / Leurs, Rob (Eds.)
Chemokine Receptors as Drug Targets
2011
ISBN: 978-3-527-32118-6
Vol. 46
Ghosh, Arun K. (Ed.)
Aspartic Acid Proteases as Therapeutic Targets
2010
ISBN: 978-3-527-31811-7
Vol. 45
Ecker, Gerhard F. / Chiba, Peter (Eds.)
Transporters as Drug Carriers
Structure, Function, Substrates
2009
ISBN: 978-3-527-31661-8
Vol. 44
List of Contributors
Frank H. Allen
Cambridge Crystallographic Data Centre (CCDC)
12 Union Road
Cambridge CB2 1EZ
UK
Karam B. Alsayyed Ahmed
University of North Carolina at Greensboro
Department of Chemistry & Biochemistry
Center for Drug Design
Greensboro, NC 27410
SUA
Pedro J. Ballester
European Bioinformatics Institute
Wellcome Trust Genome Campus
Hinxton, Cambridge CB10 1SD
UK
David A. Bardwell
Cambridge Crystallographic Data Centre (CCDC)
12 Union Road
Cambridge CB2 1EZ
UK
Caterina Barillari
The Institute of Cancer Research
Cancer Research UK Cancer Therapeutics Unit
15 Cotswold Road
Sutton SM2 5NG
UK
Nicholas P. Barton
GlaxoSmithKline Pharmaceuticals
New Frontiers Science Park (North)
Coldharbour Road
Harlow, Essex CM15 5AD
UK
Michael J. Bodkin
Eli Lilly Limited
Erl Wood Manor
Windlesham, Surrey GU20 6PH
UK
J. Phillip Bowen
University of North Carolina at Greensboro
Department of Chemistry & Biochemistry
Center for Drug Design
Greensboro, NC 27410
USA
and
Mercer University
College of Pharmacy and Health Sciences
Department of Pharmaceutical Sciences
3001 Mercer University Drive
Atlanta, GA 30341
USA
Nathan Brown
The Institute of Cancer Research
Cancer Research UK Cancer Therapeutics Unit
15 Cotswold Road
Sutton SM2 5NG
UK
Ian J. Bruno
Cambridge Crystallographic Data Centre (CCDC)
12 Union Road
Cambridge CB2 1EZ
UK
Mike Devereux
University of Basel
Klingelbergstrasse 80
4056 Basel
Switzerland
Peter Ertl
Novartis Institutes for BioMedical Research
Novartis Campus
4056 Basel
Switzerland
Marcus Gastreich
BioSolveIT
An der Ziegelei 79
53757 St. Augustin
Germany
Valerie J. Gillet
The University of Sheffield
Information School
Regent Court
211 Portobello
Sheffield S1 4DP
UK
Colin R. Groom
Cambridge Crystallographic Data Centre (CCDC)
12 Union Road
Cambridge CB2 1EZ
UK
Julian Hayward
Digital Chemistry Ltd
30 Kiveton Lane
Todwick, Sheffield S26 1HL
UK
John W. Liebeschuetz
Cambridge Crystallographic Data Centre (CCDC)
12 Union Road
Cambridge CB2 1EZ
UK
Nicholas A. Meanwell
Bristol-Myers Squibb Pharmaceutical Research and Development
Department of Medicinal Chemistry
5 Research Parkway
Wallingford, CT 06492
USA
David Millan
Sandwich Laboratories
Pfizer Global Research and Development
Ramsgate Road
Sandwich, Kent CT13 9NJ
UK
James E. J. Mills
Sandwich Laboratories
Pfizer Global Research and Development
Ramsgate Road
Sandwich, Kent CT13 9NJ
UK
Tjelvar J. Olsson
Cambridge Crystallographic Data Centre (CCDC)
12 Union Road
Cambridge CB2 1EZ
UK
George Papadatos
Eli Lilly Limited
Erl Wood Manor
Windlesham, Surrey GU20 6PH
UK
Paul L.A. Popelier
University of Manchester
Manchester Interdisciplinary Biocentre (MIB)
131 Princess Street
Manchester M1 7DN
UK
and
University of Manchester
School of Chemistry
Oxford Road
Manchester M13 9PL
UK
Matthias Rarey
ZBH University of Hamburg
Bundesstrasse 43
20146 Hamburg
Germany
Gisbert Schneider
ETH Zurich
Institute of Pharmaceutical Sciences
8093 Zurich
Switzerland
Jason Shanley
Abbott Laboratories
Global Pharmaceutical Research and Development
Department of Structural Biology
100 Abbott Park Road
Abbott Park, IL 60031
USA
Dennis A. Smith
Sandwich Laboratories
Pfizer Global Research and Development
Ramsgate Road
Sandwich, Kent CT13 9NJ
UK
Kent D. Stewart
Abbott Laboratories
Global Pharmaceutical Research and Development
Department of Structural Biology
100 Abbott Park Road
Abbott Park, IL 60031
USA
István Ujváry
iKem BT
Búza u. 32
1033 Budapest
Hungary
Peter Willett
University of Sheffield
Information School
Sheffield S1 4DP
UK
Chapter 1
Bioisosterism in Medicinal Chemistry
Nathan Brown
1.1 Introduction
One of the key challenges for the medicinal chemist today is the modulation and mediation of the potency of a small-molecule therapeutic against its biological target. In addition, it is essential to ensure that the molecule reaches its target effectively while also ensuring that it satisfies necessary safety requirements. One of the most significant approaches to assist in efficiently navigating the available chemistry space is that of bioisosteric replacement.
This book, the first dedicated solely to the subject of bioisosterism, covers the field from the very beginning to its development as a reliable and well-used approach to assist in drug design. This book is split into four parts. The first part covers the principles and theory behind isosterism and bioisosterism. The second part investigates methods that apply knowledge bases of experimental data from a variety of sources to assist in decision making. The third part reports on the four main computational approaches to bioisosteric identification and replacement using molecular properties, topology, shape, and protein structure. This book concludes with real-world examples of bioisosterism in application and a collection of reflections and perspectives on bioisosteric identification and replacement from many of the current leaders in the field.
This chapter provides an overview of the history of bioisosterism from its beginning in the early twentieth century to the present day. We also provide an overview of the importance of judicious bioisosteric replacement in lead optimization to assist in the path toward a viable clinical candidate and, ultimately, a drug.
1.2 Isosterism
James Moir [1] first considered isosterism in all but name, in 1909. It was not until 1919 that the term isosterism was given to this phenomenon by Irving Langmuir [2] in his landmark paper “Isomorphism, isosterism and covalence.” The focus of this early isosterism work was on the electronic configuration of atoms. Langmuir used experiment to identify the correspondence between the physical properties of different substances. Langmuir, in accordance with the octet rule where atoms will often combine to have eight electrons in their valence shells, compared the number and arrangement of electrons between nitrogen, carbon monoxide, and the cyanogen ion and identified that these would be the same. This relationship was demonstrated to be true between nitrogen and carbon monoxide in terms of their physical properties. The same similarities were also reported between nitrous oxide and carbon dioxide when taking experimental data from Landolt–Börnstein's tables and Abegg's handbook ().
Experimental data from Landolt–Börnstein's tables and Abegg's handbook for nitrous oxide (N2O) and carbon dioxide (CO2)
| Critical pressure (atm) |
75 |
77 |
| Critical temperature (°C) |
35.4 |
31.9 |
| Viscosity at 20 °C |
148 × 10−6 |
148 × 10−6 |
| Heat conductivity at 100 °C |
0.0506 |
0.0506 |
| Density of liquid at −20 °C |
0.996 |
1.031 |
| Density of liquid at + 10 °C |
0.856 |
0.858 |
| Refractive index of liquid, D line, 16 °C |
1.193 |
1.190 |
| Dielectric constant of liquid at 0 °C |
1.598 |
1.582 |
| Magnetic susceptibility of gas at 40 atm, 16 °C |
0.12 × 10−6 |
0.12 × 10−6 |
| Solubility in water at 0 °C |
1.305 |
1.780 |
| Solubility in alcohol at 15 °C |
3.25 |
3.13 |
However, Langmuir identified one distinct property that is substantially different between nitrous oxide and carbon dioxide, the freezing point: −102 and −56 °C, respectively. Evidence for this was assumed to be due to the freezing point being “abnormally sensitive to even slight differences in structure.”
With this observation of the correlation between the structure and arrangement of electrons with physical properties, Langmuir defined the neologism calling them isosteres, or isosteric compounds. Langmuir defined isosterism as follows:
“Comolecules are thus isosteric if they contain the same number and arrangement of electrons. The comolecules of isosteres must, therefore, contain the same number of atoms. The essential differences between isosteres are confined to the charges on the nuclei of the constituent atoms. Thus in carbon dioxide the charges on the nuclei of the carbon and oxygen atoms are 6 and 8, respectively, and there are 2 × 8 + 6 = 22 electrons in the molecule. In nitrous oxide the number of charges on the nitrogen nuclei is 7, but the total number of electrons in the molecule is again 2 × 7 + 8 = 22. The remarkable similarity of the physical properties of these two substances proves that their electrons are arranged in the same manner.”
The list of isosteres that Langmuir described in 1919 is given in . Langmuir extended his concept of isosterism to predicting likely crystal forms using sodium and fluorine ions as exemplars, these having been solved by William Henry Bragg and William Lawrence Bragg – father and son who were together awarded the Nobel Prize for Physics in 1915. Since the magnesium and oxygen ions are isosteric with the sodium and fluorine ions, it follows that magnesium oxide will have a crystal structure that is identical to that of sodium fluoride.
List of isosteres defined by Langmuir in 1919.
| 1 |
H−, He, Li+ |
| 2 |
O2−, F−, Ne, Na+, Mg2+, Al3+ |
| 3 |
S2−, Cl−, A, K+, Ca2+ |
| 4 |
Cu+, Zn2+ |
| 5 |
Br−, Kr, Rb+, Sr2+ |
| 6 |
Ag+, Cd2+ |
| 7 |
I−, Xe, Cs+, Ba2+ |
| 8 |
N2, CO, CN− |
| 9 |
CH4, NH4+ |
| 10 |
CO2, N2O, N3−, CNO− |
| 11 |
NO3−, CO32− |
| 12 |
NO2−, O3 |
| 13 |
HF, OH− |
| 14 |
ClO4−, SO42−, PO43− |
| 15 |
ClO3−, SO42−, PO43− |
| 16 |
SO3, PO3− |
| 17 |
S2O62−, P2O64− |
| 18 |
S2O72−, P2O74− |
| 19 |
SiH4, PH4+ |
| 20 |
MnO4−, CrO42− |
| 21 |
SeO42−, AsO43− |
In 1925, H.G. Grimm [3] extended the concept of isosterism, introduced by Langmuir, with Grimm's hydride displacement law:
“Atoms anywhere up to four places in the periodic system before an inert gas change their properties by uniting with one to four hydrogen atoms, in such a manner that the resulting combinations behave like pseudoatoms, which are similar to elements in the groups one to four places, respectively, to their right.”
Therefore, according to this law, the addition of hydrogen to an atom will result in a pseudoatom with similar properties to the atom of the next highest atomic number. So, CH is isosteric with N and NH is isosteric with O and so on.
Beginning in 1932, Friedrich Erlenmeyer [4, 5] extended the concepts from Grimm further and the first applications of isosterism to biological systems. Erlenmeyer redefined isosteres as:
“. . .elements, molecules or ions in which the peripheral layers of electrons may be considered identical.”
In addition, Erlenmeyer also proposed the following three additions to the concept of isosteres:
1. All elements within the same group in the periodic table are isosteres of each other. Therefore, silicon and carbon are isosteres of each other, as are oxygen and sulfur.
2. Pseudoatoms are included to characterize groups that appear superficially different but are actually very similar in physical properties. Pseudohalogens are an instance of this class, where Cl ≈ CN ≈ SCN, and so on.
3. Finally, ring equivalences are included to permit isosteric matches between different ring systems. One example is the isosteric properties between benzene and thiophene, where –CH=CH– ≈ –S–.
It was with Erlenmeyer that the concept of bioisosterism was introduced to differentiate from classical isosteres, ensuring its relevance to medicinal chemistry. The introduction of ring equivalences is significant. This was the formalization of what we consider to be a bioisosteric comparison and is the first definition of most relevance to medicinal chemistry.
1.3 Bioisosterism
Classical isosteres are traditionally categorized into the following distinct groupings [6]:
1. Monovalent atoms or groups.
2. Divalent atoms or groups.
3. Trivalent atoms or groups.
4. Tetrasubstituted atoms.
5. Ring equivalents.
A number of classical bioisosteric examples are provided in that illustrate typical replacements possible in each of these five groups.
Some examples of classical bioisosteres – groups in each row are equivalent.
| Monovalent bioisosteres |
| F, H |
| OH, NH |
| F, OH, NH, or CH3 for H |
| SH, OH |
| Cl, Br, CF3 |
| Divalent bioisosteres |
| –C=S, –C=O, –C=NH, –C=C– |
| Trivalent atoms or groups |
| –CH=, –N= |
| –P=, –As= |
| Tetrasubstituted atoms |
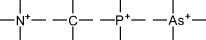 |
| Ring equivalents |
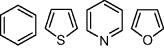 |
However, more recent definitions of isosterism, and more specifically bioisosterism, relax these constraints and permit bioisosteric pairings between moieties that do not necessarily contain the same number of atoms. Specifically, nonclassical bioisosteres include the addition of the following two groups:
1. Rings versus acyclic structures.
2. Exchangeable groups.
The origins of classical isosterism focused largely on the electronic similarity of groups rather than their functional similarity. As investigation into the field progressed, it became obvious that these very defined rules on isosterism, although powerful, were restrictive in particular to medicinal chemistry. The addition of the latter two groups for nonclassical bioisosteres permitted the mimicking of spatial arrangements, electronic properties, or another physicochemical property that is important for biological activity.
In extending and broadening the purer rules of classical isosterism, two scientists are credited with progressing the field of bioisosterism: Friedman and Thornber. In 1951, Friedman [7] provided the first definition closest to what we call bioisosterism today:
“[bioisosteres are structural moieties] which fit the broadest definition of isosteres and have the same type of biological activity.”
With this definition, the generalization of what constitutes bioisosterism was formed. However, this definition really only considers the macromolecular recognition of bioisosteres, which is of course highly important, but largely ignores the specifics of the numerous other physicochemical properties that are optimized in a medicinal chemistry project. Friedman's definition was followed in 1979 with the much less specific definition from Thornber [8] of bioisosteres and nonclassical bioisosteres:
“Bioisosteres are groups or molecules which have chemical and physical similarities producing broadly similar biological properties.”
At first reading, this definition looks somewhat similar to Friedman's, but it is the relevant importance of chemical and physical similarities that differentiates this from Friedman's definition. In addition to this definition, Thornber also defined eight parameters that could be considered in making an alteration to a structural moiety to elicit a bioisosteric pairing:
1. Size: molecular weight.
2. Shape: bond angles and hybridization states.
3. Electronic distribution: polarizability, inductive effects, charge, and dipoles.
4. Lipid solubility.
5. Water solubility.
6. pKa.
7. Chemical reactivity, including likelihood of metabolism.
8. Hydrogen bonding capacity.
Depending on the particular property that is modified by a bioisosteric replacement, the result will typically fall into one or more of the following:
1. Structural: Structural moieties often have a role in maintaining a preferred conformation and parameters such as size and bond angle play a key role in achieving this. Typically, this is particularly relevant for moieties that are embedded deep within the overall chemical structure. Scaffold hopping can be seen as an example of this, where the relative geometries of the exit vectors have a very low tolerance to modification.
2. Receptor interactions: When the moiety that is being replaced interacts directly with a receptor or enzyme, then the most relevant parameters will be size, shape, electronic properties, pKa, chemical reactivity, and hydrogen bonding.
3. Pharmacokinetics: Quite often during and after optimization of the direct biological response, it will be important to also optimize the absorption, transport, and excretion properties of the molecule. In these situations, the most important parameters to consider are lipophilicity, hydrophilicity, hydrogen bonding, and pKa.
4. Metabolism: A particular moiety may be involved in blocking or assisting with metabolism. Chemical reactivity is therefore an important property to optimize. Thornber gave the example of chloro and methyl groups on benzene being potentially interchangeable for some situations. However, the toluene derivative could be metabolized to a benzoic acid with the result being a short half-life or unexpected side effects.
These four key generalized parameters, with specific properties governing the optimization of each, provide what can be formalized as the changes that may be made in lead optimization to provide guidance on the optimization of functional groups that are bioisosteric.
In 1991, Alfred Burger [9] defined bioisosterism as:
“Compounds or groups that possess near-equal molecular shapes and volumes, approximately the same distribution of electrons, and which exhibit similar physicochemical properties. . .”
Burger's definition succinctly defines bioisosteres including all of the aforementioned extensions defined by other scientists in the field. The next section focuses on the specific improvements in lead optimization that can be gained by prudent application of the concepts of bioisosterism.
1.4 Bioisosterism in Lead Optimization
One of the processes where bioisosteric replacement can have a substantial impact, particularly in the discovery of a novel small-molecule therapeutic, is in the lead optimization stage of a drug discovery project. Once a lead molecule has been identified, the medicinal chemist is faced with the considerable challenge of making small, defined changes to an identified core structure (also chemotype or scaffold) by the addition or substitution of functional groups to test specific hypotheses. While the challenge of scaffold hopping (the replacement of the functional or specific exit geometries of a molecular scaffold) is important, this challenge will only be considered as a subset of bioisosteric replacement in this book [14–18].
1.4.1 Common Replacements in Medicinal Chemistry
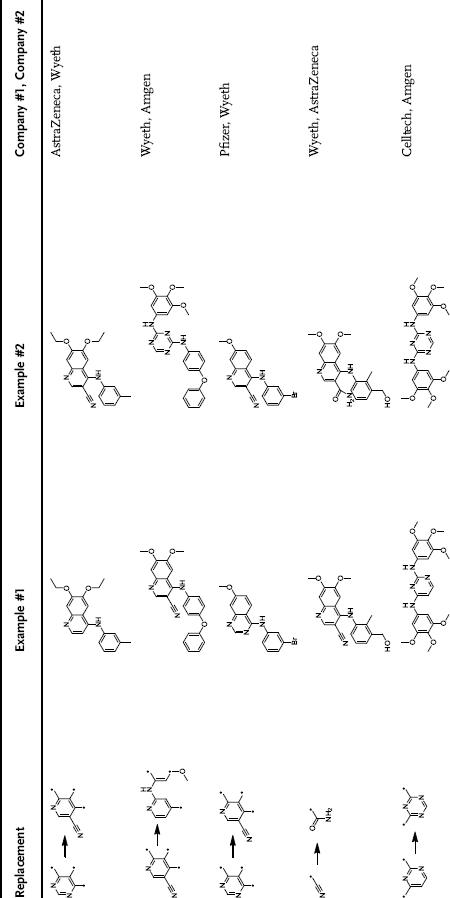
When considering a medicinal chemistry project where a lead molecule has been identified, and also chemical handles, to permit the synthesis of many analogues, the project team will identify substituents that are potential bioisosteric replacements using a number of different methods. Many of these methods will be discussed in Parts Two and Three of this book from the literature and in silico modeling approaches, respectively. Southall and Ajay [10] reported a number of common medicinal chemistry bioisosteric replacements from kinase drug candidates (). Sildenafil (Viagra)  Vardenafil (Levitra) [PDE5 Inhibitor: Pfizer Bayer AG, SP, GSK] Ciprofloxacin (Proquin) Levofloxacin (Tavanic) [Antibacterial: Bayer AG Sanofi-Aventis] Gefitinib (Iressa) Erlotinib (Tarceva) [EGFR Inhibitor: AZ Roche/ISI].
Vardenafil (Levitra) [PDE5 Inhibitor: Pfizer Bayer AG, SP, GSK] Ciprofloxacin (Proquin) Levofloxacin (Tavanic) [Antibacterial: Bayer AG Sanofi-Aventis] Gefitinib (Iressa) Erlotinib (Tarceva) [EGFR Inhibitor: AZ Roche/ISI].
Common replacements in medicinal chemistry taken from the literature [10]

1.4.2 Structure-Based Drug Design
It is becoming increasingly common that protein–ligand cocrystal structures are available to assist early on in a drug design project. The inclusion of structural information allows the design of molecules that take into account what may or may not be tolerated in a particular position, according to the conformations of key protein structure residues. This is in contrast to only using the information within the ligands that have already been synthesized and tested. The latter can lead to the assumption that the bioisosteric replacement must have the same bulk properties as the original group or, more frequently, lead to inefficiency in the design process through the unnecessary synthesis of molecules that function only to probe functional group tolerability at different positions on a molecule.
The application of protein structures to suggest bioisosteric replacements will be covered more fully in Chapter 10.
1.4.3 Multiobjective Optimization
As has been discussed previously, lead optimization involves the separate, although sometimes simultaneous, optimization of multiple parameters. When considering replacement of key functional groups around a common molecular scaffold, the chemical space of potential molecules that could be synthesized (assuming no issues in terms of synthetic accessibility, stability, etc.) is the product of the number of feasible replacement groups at each substitution point on the molecular scaffold. For example, a project with one chemical scaffold that has three points of variation, using a conservative set of 50 possible monomers at each substitution point, generates a potential project chemical space (i.e., the set of all molecules that could be synthesized) of 125 000. Typically, a medicinal chemistry project can only realize the synthesis of a small proportion of these virtual compounds, for example, approximately 1%. Therefore, the design of which molecules to synthesize and test is of great importance to ensure that those molecules are most likely to fulfill the design objectives.
To effectively and efficiently propose the most appropriate molecules for synthesis, two key points should be considered by the project team: exploration and exploitation. Exploration uses a molecular diversity measure to efficiently cover the space of virtual molecules with an even distribution of known properties. This leads to a high confidence that the entirety of the space is represented with as few molecules as necessary to demonstrate regions of specific interest. This can be achieved using a wide variety of diversity selection algorithms [11]. Here, the question being asked is that of the entirety of the chemical space.
The coverage of diversity must also be balanced with the synthesis of very close analogues to finesse those properties that are important for that specific project, many of which have been defined already in this chapter. Here, the investigation is directed on small and specific changes, most often a number of single alterations that enhance the understanding of the local structure–activity relationship (SAR). It is with this part of the lead optimization process that bioisosteric replacements are most important, as opposed to the diversity design where bioisosteric replacements will not necessarily provide sufficient information about the global chemical space [13].
Bioisosteric replacement is often considered when the aims are to maintain enzyme potency while optimizing additional properties, such as cellular penetration, solubility, metabolism, toxicity, and so on. This principle is often referred to as multiobjective optimization (MOOP) or multiparameter optimization (MPO) [12]. There are many ways in which one can address multiple objectives, but it is important to understand the landscape of the trade-off surface between each of the important objectives, including an understanding of parameters that may be correlated with each other ().
The combination of identifying bioisosteric replacements in a lead molecule together with the multiobjective prioritization of virtual molecules in that chemical series for synthesis provides the medicinal chemist with the key information for making design decisions in a therapeutic project. The approaches to identifying these replacements will be covered in Parts Two and Three of this book, but they can all be applied in this challenge.
1.5 Conclusions
The origins of isosterism have been traced back to the early twentieth century, most notably in the work of Langmuir, which also gave the concept its name. The extension of isosterism through Grimm and Erlenmeyer paved the way to the definition of bioisosterism, largely promulgated by Friedman and Thornber. Moving from a definition of isosterism that focused specifically on the electronic makeup of isosteres to a more functional outlook in terms of biological properties was a major step forward toward what we today call bioisosterism.
Bioisosterism is now one of the most important tools that medicinal chemists have at their disposal. Through shrewd application of bioisosteres that have experimental precedent or have been identified by theoretical calculations, the medicinal chemist is now well prepared with highly effective tools that have been demonstrated to be of great utility in therapeutic design programs. The remaining chapters in this part will detail the key theories behind bioisosteres and their replacement.
References
1. Meanwell, N.A. (2011) Synopsis of some recent tactical application of bioisosteres in drug design. Journal of Medicinal Chemistry, 54, 2529–2591.
2. Langmuir, I. (1919) Isomorphism, isosterism and covalence. Journal of the American Chemical Society, 41, 1543–1559.
3. Grimm, H.G. (1925) On construction and sizes of non-metallic hydrides. Zeitschrift fur Elektrochemie und Angewandte Physikalische Chemie, 31, 474; 1928, 34, 430; 1934, 47, 553–594.
4. Erlenmeyer, H. and Berger, E. (1932) Studies on the significance of structure of antigens for the production and the specificity of antibodies. Biochemical Zoology, 252, 22–36.
5. Erlenmeyer, H., Berger, E., and Leo, M. (1933) Relationship between the structure of antigens and the specificity of antibodies. Helvetica Chimica Acta, 16, 733–738.
6. Lemke, T.L. and Williams, D.A. (2007) Foye's Principles of Medicinal Chemistry, 6th edn, Lippincott Williams & Wilkins, Baltimore, MD.
7. Friedman, H.L. (1951) Influence of isosteric replacements upon biological activity. NAS-NRS Publication No. 206, NAS-NRS, Washington, DC, pp. 295–358.
8. Thornber, C.W. (1979) Isosterism and molecular modification in drug design. Chemical Society Reviews, 8, 563–580.
9. Burger, A. (1991) Isosterism and bioisosterism in drug design. Progress in Drug Research, 37, 288–362.
9. Southall, N.T. and Ajay (2006) Kinase patent space visualization using chemical replacements. Journal of Medicinal Chemistry, 49, 2103–2109.
11. Gillet, V.J. (2011) Diversity selection algorithms. Wiley Interdisciplinary Reviews: Computational Molecular Science, 1, 580–589.
12. Nicolaou, C.A., Brown, N., and Pattichis, C.K. (2007) Molecular optimization using multi-objective methods. Current Opinion in Drug Discovery & Development, 10, 316–324.
13. Brown, N. and Lewis, R.A. (2006) Exploiting QSAR methods in lead optimization. Current Opinion in Drug Discovery & Development, 9, 419–424.
14. Ciapetti, P. and Giethlen, B. (2008) Molecular variations based on isosteric replacements, in The Practice of Medicinal Chemistry, 3rd edn (ed. C.G. Wermuth), Elsevier.
15. DiMasi, J. and Faden, L.B. (2011) Competitiveness in follow-on drug R&D: a race of imitation? Nature Reviews Drug Discovery, 10, 23–27.
16. Langdon, S.R., Ertl, P., and Brown, N. (2010) Bioisosteric replacement and scaffold hopping in lead generation and optimization. Molecular Informatics, 29, 366–385.
17. Patani, G.A. and LaVoie, E.J. (1996) Bioisosterism: a rational approach in drug design. Chemical Reviews, 96, 3147–3176.
18. Wermuth, C.G. (2006) Similarity in drugs: reflections on analogue design. Drug Discovery Today, 11, 348–354.


