Table of Contents
Cover
Dedication
Title page
Copyright page
Preface
How to get the best out of your textbook
The Anytime, Anywhere Textbook
Instructors … receive your own digital desk copies!
Features contained within your textbook
Part One: Basic Principles of Human Genetics
CHAPTER 1 DNA Structure and Function
Deoxyribonucleic Acid
Gene Function
Conclusion
CHAPTER 2 Genetic Variation
DNA Sequence Variants
Causes of Nucleotide Mutation
Gene Duplication and Evolution
Genetic Polymorphism
Conclusion
CHAPTER 3 Patterns of Inheritance
Mendelian Inheritance
Mitochondrial Inheritance
Conclusion
CHAPTER 4 The Human Genome
Gene Cloning
Gene Mapping
The Human Genome
Conclusion
CHAPTER 5 Multifactorial Inheritance
Concept of Multifactorial Inheritance
Genetics of Common Disease
Conclusion
CHAPTER 6 Cell Division and Chromosomes
Cell Division
Chromosomes
Chromosome Abnormalities
Indications for Chromosomal Analysis
Conclusion
CHAPTER 7 Population Genetics
Hardy–Weinberg Equilibrium
Deviations from the Hardy–Weinberg Equilibrium
Conclusion
CHAPTER 8 Cancer Genetics
Cancer Is a Genetic Disorder
Tumor Suppressor Genes
Oncogenes
Normal Roles of Tumor Suppressor Genes and Oncogenes
Epigenetics and MicroRNAs in Cancer
The Molecular Basis of Oncogenesis
New Treatments for Cancer
Conclusion
Part Two: Genetics and Genomics in Medical Practice
CHAPTER 9 Chromosome Translocation
Part I
Part II
Part III
Part IV
Part V
Part VI
Clinical Cytogenetics
CHAPTER 10 Molecular Diagnosis
Part I
Part II
Part III
Part IV
Part V
Part VI
Part VII
Molecular Diagnosis of Genetic Disorders
CHAPTER 11 Newborn Screening
Part I: December 1965
Part II: December 1965
Part III: April 1973
Part IV: March 1986
Part V: September 1988
Part VI: April 2010
Inborn Errors of Metabolism
CHAPTER 12 Developmental Genetics
Part I
Part II
Part III
Part IV
Part V
Part VI
Inborn Errors of Development
CHAPTER 13 Carrier Screening
Part I
Part II
Part III
Part IV
Part V
Carrier Screening
CHAPTER 14 Genetic Risk Assessment
Part I
Part II
Part III
Part IV
Part V
Genetic Screening for Disease Risk
CHAPTER 15 Genetic Testing for Risk of Cancer
Part I
Part II
Part III
Part IV
Part V
Part VI
Part VII
Part VIII
Part IX
Familial Predisposition to Cancer
CHAPTER 16 Pharmacogenetics
Part I
Part II
Part III
Part IV
Part V
Part VI
Pharmacogenetics and Pharmacogenomics
CHAPTER 17 Treatment of Genetic Disorders
PART I
PART II
PART III
PART IV
PART V
PART VI
Therapy of Genetic Disorders
Answers to Review Questions
Glossary
Index
Access to accompanying material
Dedication
To Shelley, Jessica, and Katie – Bruce Korf
To Mallory Lynn and David Nicholas – Mira Irons
This new edition is also available as an e-book.
For more details, please see
www.wiley.com/buy/9780470654477
or scan this QR code:


This edition first published 2013 © 2013 by John Wiley & Sons, Ltd.
Previous edition: © 2007 by Bruce R. Korf, MD, PhD
Wiley-Blackwell is an imprint of John Wiley & Sons, formed by the merger of Wiley’s global Scientific, Technical and Medical business with Blackwell Publishing.
Registered office: John Wiley & Sons, Ltd, The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK
Editorial offices: 9600 Garsington Road, Oxford, OX4 2DQ, UK
The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK
111 River Street, Hoboken, NJ 07030-5774, USA
For details of our global editorial offices, for customer services and for information about how to apply for permission to reuse the copyright material in this book please see our website at www.wiley.com/wiley-blackwell.
The right of the author to be identified as the author of this work has been asserted in accordance with the UK Copyright, Designs and Patents Act 1988.
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs and Patents Act 1988, without the prior permission of the publisher.
Designations used by companies to distinguish their products are often claimed as trademarks. All brand names and product names used in this book are trade names, service marks, trademarks or registered trademarks of their respective owners. The publisher is not associated with any product or vendor mentioned in this book. This publication is designed to provide accurate and authoritative information in regard to the subject matter covered. It is sold on the understanding that the publisher is not engaged in rendering professional services. If professional advice or other expert assistance is required, the services of a competent professional should be sought.
Library of Congress Cataloging-in-Publication Data
Korf, Bruce R.
Human genetics and genomics / Bruce R. Korf, Mira B. Irons. – 4th ed.
p. ; cm.
Includes bibliographical references and index.
ISBN 978-0-470-65447-7 (pbk. : alk. paper)
I. Irons, Mira B. II. Title.
[DNLM: 1. Genetics, Medical–Problems and Exercises. 2. Genetic Diseases, Inborn–Problems and Exercises. 3. Genomics–Problems and Exercises. 4. Problem-Based Learning–Problems and Exercises.
QZ 18.2]
616'.042–dc23
2012024773
A catalogue record for this book is available from the British Library.
Wiley also publishes its books in a variety of electronic formats. Some content that appears in print may not be available in electronic books.
Cover image and design by Fortiori design
Preface
Sixteen years have passed since the first edition of this book was published, and this has been a time of enormously rapid change and advancement in the field of genetics and genomics. The increasing prominence of genomics was recognized in the last edition, when the name was changed to Human Genetics and Genomics. That edition also launched a reorganization of the book, first presenting basic concepts and then using a problem-based approach to illustrate clinical applications. That pattern has been retained in this new edition.
A major change in this fourth edition is the addition of a co-author, Dr. Mira Irons. This reflects the reality that the scope and pace of change in our field have expanded to the point where it is difficult for any one individual to keep up. Dr. Irons has rewritten or updated several of the chapters and case studies. Another change is the addition of a section called “Sources of Information” to each chapter. Genetics and genomics are intensively information-based sciences, and therefore are heavily dependent on access to continuously updated sources of information. We hope that this will help to guide students to reliable data sources that will assist in their future clinical or research activities.
In the previous edition of this book, it was noted that genomic approaches were beginning to inform our understanding of common as well as rare disorders. This trend has continued, with the advent of genome-wide association studies, and has spawned clinical applications, including direct-to-consumer genetic risk assessment. This edition has been updated to highlight these developments. There has also been remarkable progress in the approach to rare “single-gene” disorders. The list of conditions for which genetic testing is available has expanded dramatically, and recently whole-exome or whole-genome sequencing has been applied to solving diagnostic challenges. Most notably, many rare genetic conditions previously thought to be untreatable are now the target of clinical trials for new therapeutic approaches. We have also highlighted this development, along with the need to appreciate the process of performing clinical trials for genetic disorders.
As with previous editions, we have benefited greatly from comments and suggestions from both reviewers and readers. “Mutations” inevitably are found within the text, and we are most grateful to those who point these out so that they can be corrected. Likewise, we greatly appreciate suggestions for new content or better ways to explain some of the more challenging concepts.
As genomic approaches pervade all of medicine, one of the most common concerns expressed is that health providers cannot keep up with this new discipline. We remain optimistic that a new generation of students will embrace the new opportunities afforded by genetics and genomics to transform our approach to both rare and common conditions as they begin a process of learning that will span their entire careers.
Bruce R. Korf
Mira B. Irons
How to Get the Best Out of Your Textbook
Welcome to the new edition of Human Genetics and Genomics. Over the next two pages you will be shown how to make the most of the learning features included in the textbook.
The Anytime, Anywhere Textbook
For the first time, your textbook comes with free access to a Wiley E-Text: Powered by VitalSource – a digital, interactive version of this textbook which you own as soon as you download it.
Your Wiley E-Text: Powered by VitalSource allows you to:
Search: Save time by finding terms and topics instantly in your book, your notes, even your whole library (once you’ve downloaded more textbooks)
Note and Highlight: Colour code, highlight and make digital notes right in the text so you can find them quickly and easily
Organize: Keep books, notes and class materials organized in folders inside the application
Share: Exchange notes and highlights with friends, classmates and study groups
Upgrade: Your textbook can be transferred when you need to change or upgrade computers
Link: Link directly from the page of your interactive textbook to all of the material contained on the companion website
To access your Wiley E-Text: Powered by VitalSource:
- Find the redemption code on the inside front cover of this book and carefully scratch away the top coating of the label.
- Visit www.vitalsource.com/software/bookshelf/downloads to download the Bookshelf application.
- If you have puchased this title as an e-book, access to your Wiley E-Text: Powered by VitalSource is available with proof of purchase within 90 days. Visit http://support.wiley.com to request a redemption code via the “Live Chat” or “Ask A Question” tabs.
- Open the Bookshelf application on your computer and register for an account.
- Follow the registration process and enter your redemption code to download your digital book.
- For full access instructions, visit www.korfgenetics.com.
CourseSmart gives you instant access (via computer or mobile device) to this Wiley-Blackwell eTextbook and its extra electronic functionality, at 40% off the recommended retail print price. See all the benefits at www.coursesmart.com/students.
Instructors … Receive Your Own Digital Desk Copies!
It also offers instructors an immediate, efficient, and environmentally-friendly way to review this textbook for your course.
For more information visit www.coursesmart.com/instructors.
With CourseSmart, you can create lecture notes quickly with copy and paste, and share pages and notes with your students. Access your Wiley CourseSmart digital textbook from your computer or mobile device instantly for evaluation, class preparation, and as a teaching tool in the classroom.
Simply sign in at http://instructors.coursesmart.com/bookshelf to download your Bookshelf and get started. To request your desk copy, hit “Request Online Copy” on your search results or book product page.
A companion website
Your textbook is also accompanied by a FREE companion website that contains:

- Figures from the book in PowerPoint format
- Interactive self-assessment MCQ tests
- Further online reading list
- Interactive problem-based learning cases
- Factsheets on basic conditions
Log on to www.korfgenetics.com to find out more.
Features Contained within Your Textbook
Every chapter begins with a list of key points contained within the chapter and an introduction to of the topic.
‘Ethical implications’, ‘Hot topic’ and other boxes give further insight into topics.
Self-assessment review questions help you test yourself after each chapter.
Your textbook is full of clinical photographs, method notes, cases illustrations and tables. The Wiley E-Text: Powered by VitalSource version of your textbook will allow you to copy and paste any photograph or illustration into assignments, presentations and your own notes.
We hope you enjoy using your new textbook. Good luck with your studies!
Part One
Basic Principles of Human Genetics
CHAPTER 1
DNA Structure and Function
Introduction
The 20th century will likely be remembered by historians of biological science for the discovery of the structure of DNA and the mechanisms by which information coded in DNA is translated into the amino acid sequence of proteins. Although the story of modern human genetics begins about 50 years before the structure of DNA was elucidated, we will start our exploration here. We do so because everything we know about inheritance must now be viewed in the light of the underlying molecular mechanisms. We will see here how the structure of DNA sets the stage both for its replication and for its ability to direct the synthesis of proteins. We will also see that the function of the system is tightly regulated, and how variations in the structure of DNA can alter function. The story of human genetics did not begin with molecular biology, and it will not end there, as knowledge is now being integrated to explain the behavior of complex biological systems. Molecular biology, however, remains a key engine of progress in biological understanding, so it is fitting that we begin our journey here.
Key Points
- DNA consists of a double-helical sugar–phosphate structure with the two strands held together by hydrogen bonding between adenine and thymine or cytosine and guanine bases.
- DNA replication involves local unwinding of the double helix and copying a new strand from the base sequence of each parental strand. Replication proceeds bidirectionally from multiple start sites in the genome.
- DNA is complexed with proteins to form a highly compacted chromatin fiber in the nucleus.
- Genetic information is copied from DNA into messenger RNA (mRNA) in a highly regulated process that involves activation or repression of individual genes.
- mRNA molecules are extensively processed in the nucleus, including removal of introns and splicing together of exons, prior to export to the cytoplasm for translation into protein.
- The base sequence of mRNA is read in triplet codons to direct the assembly of amino acids into protein on ribosomes.
- Some genes are permanently repressed by epigenetic marks such as methylation of cytosine bases. These include most genes on one of two X chromosomes in cells in females and one of the two copies of imprinted genes.
Deoxyribonucleic Acid
Mendel described dominant and recessive inheritance before the concept of the gene was introduced and long before the chemical basis of inheritance was known. Cell biologists during the late 19th and early 20th centuries had established that genetic material resides in the cell nucleus and DNA was known to be a major chemical constituent. As the chemistry of DNA came to be understood, for a long time it was considered to be too simple a molecule – consisting of just four chemical building blocks, the bases adenine, guanine, thymine, and cytosine, along with sugar and phosphate – to account for the complexity of genetic transmission. Credit for recognition of the role of DNA in inheritance goes to the landmark experiments by Oswald Avery and his colleagues, who demonstrated that a phenotype of smooth or rough colonies of the bacterium Pneumococcus could be transmitted from cell to cell through DNA alone. Elucidation of the structure of DNA by James Watson and Francis Crick in 1953 opened the door to understanding the mechanisms whereby this molecule functions as the agent of inheritance (Sources of Information 1.1).

Sources of Information 1.1 Mendelian Inheritance in Man
Dr. Victor McKusick and his colleagues at Johns Hopkins School of Medicine began to catalog genes and human genetic traits in the 1960s. The first edition of the catalog Mendelian Inheritance in Man was published in 1966. Multiple print editions subsequently appeared, and now the catalog is maintained on the Internet as “Online Mendelian Inheritance in Man” (OMIM), located at www.omim.org.
OMIM is recognized as the authoritative source of information about human genes and genetic traits. The catalog can be searched by gene, phenotype, gene locus, and many other features. The catalog provides a synopsis of the gene or trait, including a summary of clinical features associated with mutations. There are links to other databases, providing access to gene and amino acid sequences, mutations, and so on. Each entry has a unique six-digit number, the MIM number. Autosomal dominant traits have entries beginning with 1, recessive traits with 2, X linked with 3, and mitochondrial with 5. Specific genes have MIM numbers that start with 6.
Throughout this book, genes or genetic traits will be annotated with their corresponding MIM number to remind the reader that more information is available on OMIM and to facilitate access to the site.
DNA Structure
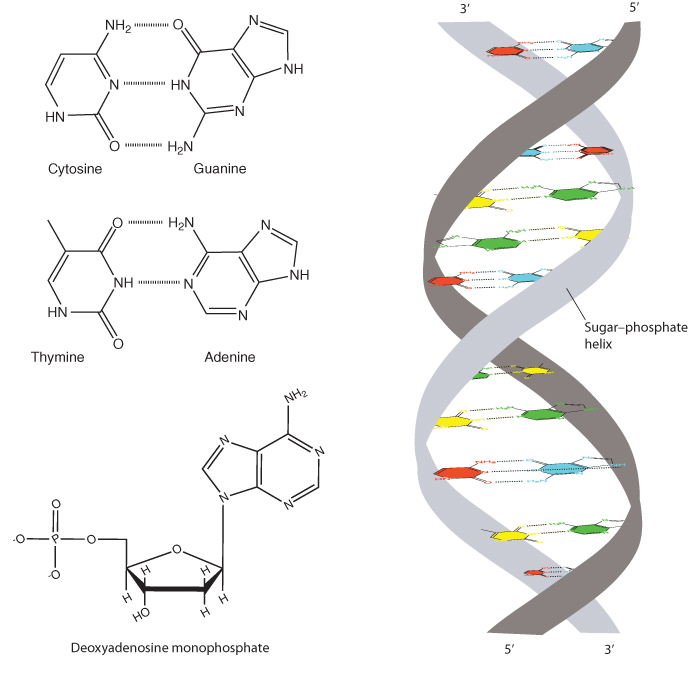
DNA consists of a pair of strands of a sugar–phosphate backbone attached to a set of pyrimidine and purine bases (Figure 1.1). The sugar is deoxyribose – ribose missing an oxygen atom at its 2′ position. Each DNA strand consists of alternating deoxyribose molecules connected by phosphodiester bonds from the 5′ position of one deoxyribose to the 3′ position of the next. The strands are bound together by hydrogen bonds between adenine and thymine bases and between guanine and cytosine bases. Together these strands form a right-handed double helix. The two strands run in opposite (antiparallel) directions, so that one extends from 5′ to 3′ while the other goes from 3′ to 5′.
The key feature of DNA, wherein resides its ability to encode information, is in the sequence of the four bases (Methods 1.1). The number of adenine bases (A) always equals the number of thymines (T), and the number of cytosines (C) always equals the number of guanines (G). This is because A on one strand is always paired with T on the other, and C is always paired with G. The pairing is noncovalent, due to hydrogen bonding between complementary bases. G–C base pairs form three hydrogen bonds, whereas A–T pairs form two, making the G–C pairs slightly more thermodynamically stable. Because the pairs always include one purine base (A or G) and one pyrimidine base (C or T), the distance across the helix remains constant.

Methods 1.1 Isolation of DNA
DNA, or in some cases RNA, is the starting point for most experiments aimed at studying gene structure or function. DNA can be isolated from any cell that contains a nucleus. The most commonly used tissue for human DNA isolation is peripheral blood, where white blood cells provide a readily accessible source of nucleated cells. Other commonly used tissues include cultured skin fibroblasts, epithelial cells scraped from the inner lining of the cheek, and fetal cells obtained by amniocentesis or chorionic villus biopsy. Peripheral blood lymphocytes can be transformed with Epstein–Barr virus into immortalized cell lines, providing permanent access to growing cells from an individual.
Nuclear DNA is complexed with proteins, which must be removed in order for the DNA to be analyzed. For some experiments it is necessary to obtain highly purified DNA, which involves digestion or removal of the proteins. In other cases, relatively crude preparations suffice. This is the case, for example, with DNA isolated from cheek scrapings. The small amount of DNA isolated from this source is usually released from cells with minimal effort to remove proteins. This preparation is adequate for limited analysis of specific gene sequences. DNA preparations can be obtained from very minute biological specimens, such as drops of dried blood, skin cells, or hair samples isolated from crime scenes for forensic analysis.
Isolation of RNA involves purification of nucleic acid from the nucleus and/or cytoplasm. This RNA can be used to study the patterns of gene expression in a particular tissue. RNA tends to be less stable than DNA, requiring special care during isolation to avoid degradation.
DNA Replication
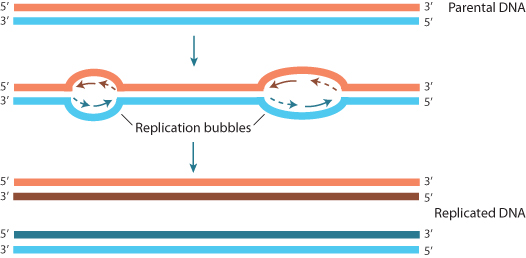
The complementarity of A to T and G to C provides the basis for DNA replication, a point that was recognized by Watson and Crick in their paper describing the structure of DNA. DNA replication proceeds by a localized unwinding of the double helix, with each strand serving as a template for replication of a new sister strand (Figure 1.2). Wherever a G base is found on one strand, a C will be placed on the growing strand; wherever a T is found, an A will be placed; and so on. Bases are positioned in the newly synthesized strand by hydrogen bonding, and new phosphodiester bonds are formed in the growing strand by the enzyme DNA polymerase. This is referred to as semiconservative replication, because the newly synthesized DNA double helices are hybrid molecules that consist of one parental strand and one new daughter strand. Unwinding of the double helix is accomplished by another enzyme system, helicase.
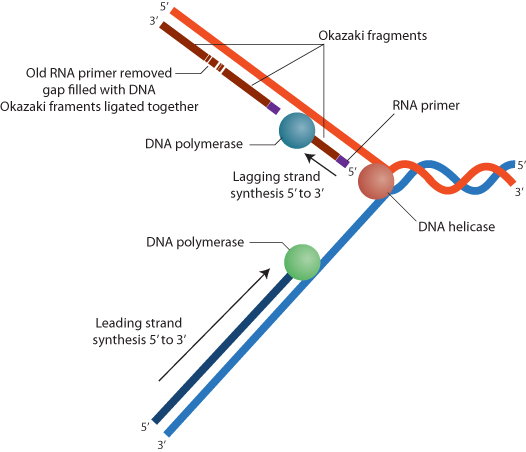
DNA replication requires growth of a strand from a preexisting primer sequence. Primer sequences are provided by transcription of short RNA molecules from the DNA template. RNA is a single-stranded nucleic acid, similar to DNA except that the sugar molecules are ribose rather than deoxyribose and uracil substitutes for thymine (and pairs with adenine). During DNA replication, short RNA primers are transcribed and then extended by DNA polymerase. DNA is synthesized in a 5′ (exposed phosphate on the 5′ carbon of the ribose molecule) to 3′ (exposed hydroxyl on the 3′ carbon) direction. For one strand, referred to as the leading strand, this can be accomplished continuously as the DNA unwinds. The other strand, called the lagging strand, is replicated in short segments, called Okazaki fragments, which are then enzymatically ligated together by DNA ligase. Distinct polymerases replicate the leading and lagging strands. The short RNA primers are ultimately removed and replaced with DNA to complete the replication process.
The human genome consists of over 3 billion base pairs of DNA packaged into 23 pairs of chromosomes. Each chromosome consists of a single, continuous DNA molecule, encompassing tens to hundreds of millions of base pairs. If the DNA on each chromosome were to be replicated in a linear manner from one end to another, the process would go on interminably – certainly too long to sustain the rates of cell division that must occur. In fact, the entire genome can be replicated in a matter of hours because replication occurs simultaneously at multiple sites along a chromosome. These origins of replication are bubble-like structures from which DNA replication proceeds bidirectionally until an adjacent bubble is reached (Figure 1.3).
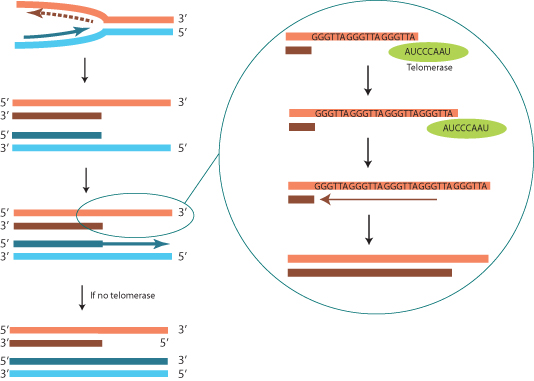
One special case in DNA replication is the replication of the ends of chromosomes. Removal of the terminal RNA primer from the lagging strand at the end of a chromosome would result in shortening of the end, since there is no upstream primer for DNA polymerase to replace the short RNA primer. This problem is circumvented by action of an enzyme called telomerase, which uses an RNA template intrinsic to the enzyme to add a stretch of DNA onto the 3′ end of the lagging strand (Figure 1.4). The DNA sequence of the telomere is determined by the RNA sequence in the enzyme; for humans the sequence is GGGTTA. Each chromosome end has a tandem repeat of thousands of copies of the telomere sequence that is replicated during early development. Somatic cells may replicate without telomerase activity, resulting in a gradual shortening of the ends of the chromosomes with successive rounds of replication. This may be one of the factors that limits the number of times a cell can divide before it dies, a phenomenon known as senescence (Clinical Snapshot 1.1).
- - - - - - - - - -

Clinical Snapshot 1.1 Dyskeratosis Congenita
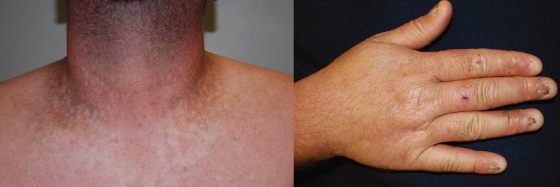
Eddy is a 4-year-old boy brought in by his parents because of recurrent cough. He has had two bouts of pneumonia that required treatment with antibiotics over the past 2 months. Now he is sick again, having never stopped coughing since the last episode of pneumonia. His parents have noted that he has had low energy over the past several weeks. His examination shows a fever of 39°C and rapid respirations with frequent coughing. His breath sounds are abnormal on the right side of his chest. He also has hyperkeratotic skin with streaky hyperpigmentation. His finger and toenails are thin and broken at the ends, and his hair is sparse. A blood count shows anemia and a reduced number of white blood cells. A bone marrow aspirate is obtained, and it shows generalized decrease in all cell lineages. A clinical diagnosis of dyskeratosis congenita is made.
Dyskeratosis congenita consists of reticulated hyperpigmentation of the skin, dystrophic hair and nails, and generalized bone marrow failure (Figure 1.5). It usually presents in childhood, often with signs of pancytopenia. There is an increased rate of spontaneous chromosome breakage seen in peripheral blood lymphocytes. Dyskeratosis congenita can be inherited as an X-linked recessive (MIM 305000), autosomal dominant (MIM 127550), or autosomal recessive (MIM 224230) trait. The X-linked form is due to mutation in a gene that encodes the protein dyskerin (MIM 300126). Dyskerin is involved in the synthesis of ribosomal RNA and also interacts with telomerase. The autosomal dominant form is due to mutation in the gene hTERC (MIM 602322). hTERC encodes the RNA component of telomerase. The autosomal recessive form can be due to mutations in NOLA2 (MIM 606470) or NOLA3 (MIM 606471); both encode proteins that interact with dyskerin. The X-linked recessive form is more severe and earlier in onset than the dominant form. Both forms are associated with defective telomere functioning, leading to shortened telomeres. This likely leads to premature cell death and also explains the spontaneous chromosome breakage. The phenotype of the X-linked form may also be due, in part, to defective rRNA processing.
- - - - - - - - - -
Chromatin
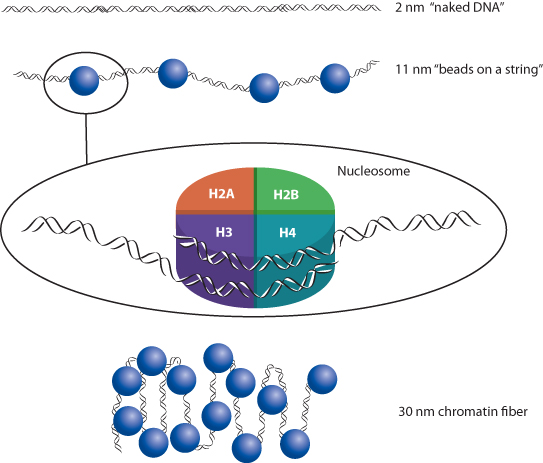
The DNA within each cell nucleus must be highly compacted to accommodate the entire genome in a very small space. The enormous stretch of DNA that comprises each chromosome is actually a highly organized structure (Figure 1.6). The DNA double helix measures approximately 2 nm in diameter, but DNA does not exist in the nucleus in a “naked” form. It is complexed with a set of lysine- and arginine-rich proteins called histones. Two molecules of each of four major histone types – H2A, H2B, H3, and H4 – associate together with about every 146 base pairs to form a structure known as the nucleosome, which results in an 11 nm thick fiber. Nucleosomes are separated from one another by up to 80 base pairs, like beads on a string. This is more or less the conformation of actively transcribed chromatin but, during periods of inactivity, some regions of the genome are more highly compacted. The next level of organization is the coiling of nucleosomes into a 30 nm thick chromatin fiber held together by another histone, H1, and other nonhistone proteins. Chromatin is further compacted into the highly condensed structures comprising each chromosome, with the maximum condensation occurring during the metaphase stage of mitosis (see Chapter 6).
Gene Function
The basic tenet of molecular genetics – often referred to as the central dogma – is that DNA encodes RNA, which in turn encodes the amino acid sequence of proteins. It is now clear that this is a simplified view of the function of the genome. As will be seen in Chapter 4, much of the DNA sequence does not encode protein. A large proportion of the genome consists of noncoding sequences, such as repeated DNA, or encodes RNA that is not translated into protein. Nevertheless, the central dogma remains a critical principle of genome function. We will explore here the flow of information from DNA to RNA to protein.
Transcription
The process of copying the DNA sequence of a gene into messenger RNA (mRNA) is referred to as transcription. Some genes are expressed nearly ubiquitously. These are referred to as housekeeping genes. They include genes necessary for cell replication or metabolism. For other genes, expression is tightly controlled, with particular genes being turned on or off in particular cells at specific times in development or in response to physiological signals. Genomic studies are now being applied to analysis of gene regulation and are revealing remarkable details on the structure and function of regulatory elements in the human genome (Hot Topic 1.1).

Hot Topic 1.1 Encode Project
The sequencing of the human genome (see Chapter 2) has made it possible to identify the full complement of genes, but does not in itself reveal how these genes are regulated. In 2003 a project was launched to characterize the entire set of coding elements in the genome, including identification of all genes and their corresponding regulatory sequences. The project is referred to as ENCODE, which is an acronym for encyclopedia of DNA elements. A pilot study was published in 2007 and more complete results were published in a series of papers in 2012.
The project has revealed a number of surprises. A total of 20 687 protein-encoding genes (not representing the full complement) were identified, each with, on average, 6.3 alternatively spliced transcripts. Among RNAs that do not encode protein, there were 8 801 small RNAs and 9 640 long non-coding transcripts. More than 80% of the genome was found to be transcribed into some kind of RNA in at least one cell type, a much higher level of transcriptional activity than expected. Ninety-five percent of the genome was found to be within 8 kb of a region involved in DNA/protein interaction. Nearly 400 000 regions were found to have features of enhancers and over 70 000 had features of promoter activity. Genetic variants associated with common disease (see Chapter 5) were found to be enriched in regions with functional elements that do not encode protein. It has become clear that much of the genome is dedicated to encoding elements responsible for fine control of gene expression in distinct cell types.
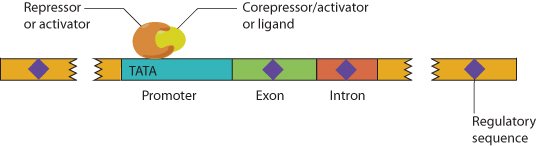
Gene expression is regulated by proteins that bind to DNA and either activate or repress transcription. The anatomy of elements that regulate gene transcription is shown in Figure 1.7. The promoter region is immediately adjacent to the transcription start site, usually within 100 base pairs. Most promoters include a base sequence of T and A bases called the TATA box. In some cases there may be multiple, alternative promoters at different sites in a gene that respond to regulatory factors in different tissues. Regulatory sequences may occur adjacent to the promoter or may be located thousands of base pairs away. These distantly located regulatory sequences are known as enhancers. Enhancer sequences function regardless of their orientation with respect to the gene.
DNA-binding proteins may serve as repressors or activators of transcription, and may bind to the promoter, to upstream regulatory regions, or to more distant enhancers. Activator or repressor proteins are regulated by binding of specific ligands. Ligand binding changes the confirmation of the transcription factor and may activate it or inactivate it. The ligand is typically a small molecule, such as a hormone. Many transcription factors form dimers, either homodimers of two identical proteins or heterodimers of two different proteins. There may also be corepressor or coactivator proteins. Some transcription factors stay in the cytosol until the ligand binds or some other activation process occurs, at which time they move to the nucleus to activate their target gene(s). In other situations, the transcription factors reside in the nucleus most of the time and may even be located at the response element sequences, but without the ligand they are inactive or even repress transcription.
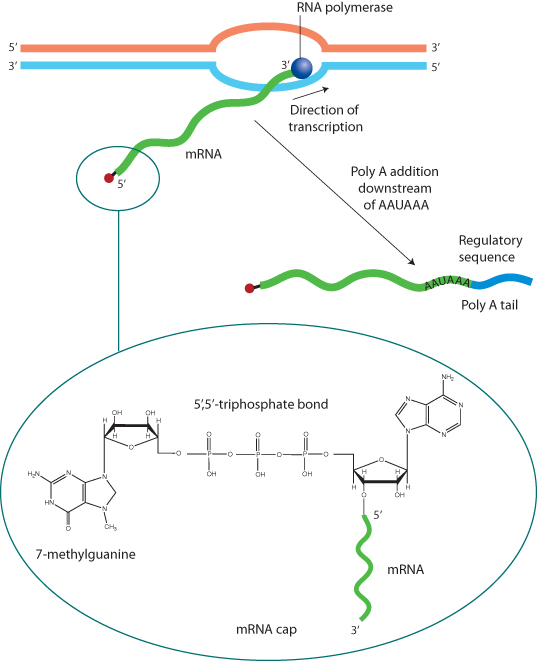
Transcription begins with the attachment of the enzyme RNA polymerase to the promoter (Figure 1.8). There are three major types of RNA polymerase, designated types I, II, and III. Most gene transcription is accomplished by RNA polymerase II. Type I is involved in the transcription of ribosomal RNA (rRNA), and type III transcribes transfer RNA (tRNA) (see the “Translation” section of this chapter). The polymerase reads the sequence of the DNA template strand, copying a complementary RNA molecule, which grows from the 5′ to the 3′ direction. The resulting mRNA is an exact copy of the DNA sequence, except that uracil takes the place of thymine in RNA. Soon after transcription begins, a 7-methyl guanine residue is attached to the 5′-most base, forming the cap. Transcription proceeds through the entire coding sequence. Some genes include a sequence near the 3′ end that signals RNA cleavage at that site and enzymatic addition of 100 to 200 adenine bases, the poly-A tail. Polyadenylation is characteristic of housekeeping genes, which are expressed in most cell types. Both the 5′ cap and the poly-A tail stabilize the mRNA molecule and facilitate its export to the cytoplasm.
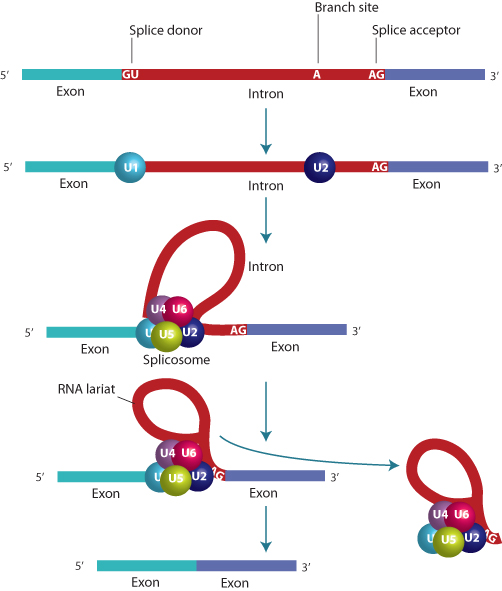
The DNA sequence of most genes far exceeds the length required to encode their corresponding proteins. This is accounted for by the fact that the coding sequence is broken into segments, called exons, which are interrupted by noncoding segments called introns. Some exons may be less than a hundred bases long, whereas introns can be several thousand bases in length; therefore, much of the length of a gene may be devoted to introns. The number of exons in a gene may be as few as one or two, or may number in the dozens. The processing of the transcript into mature mRNA requires the removal of the introns and splicing together of the exons (Figure 1.9), carried out by an enzymatic process that occurs in the nucleus. The 5′ end of an intron always consists of the two bases GU, followed by a consensus sequence that is similar, but not identical, in all introns. This is the splice donor. The 3′ end, the splice acceptor, ends in AG, preceded by a consensus sequence.
The splicing process requires complex machinery composed of both proteins and small RNA molecules (small nuclear RNA, or snRNA), consisting of fewer than 200 bases. snRNA is also transcribed by RNA polymerase II. The splice is initiated by binding of a protein–RNA complex to the splice donor, at a point within the intron called the branch point, and the splice acceptor. First the RNA is cleaved at the donor site and this is attached in a 5′–2′ bond to the branch point. Then the acceptor site is cleaved, releasing a lariat structure that is degraded, and the 5′ and 3′ ends are ligated together. The splicing process also requires the function of proteins, SR proteins, which are involved in selecting sites for the initiation of splicing. These proteins interact with sequences known as splice enhancers or silencers. The splicing process is vulnerable to disruption by mutation, as might be predicted from its complexity (see Chapter 4).
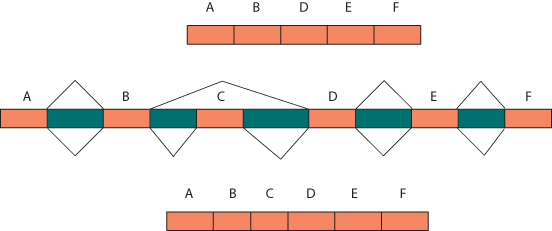
The RNA-splicing process offers a point of control of gene expression. Under the influence of control molecules present in specific cells, particular exons may be included or not included in the mRNA due to differential splicing (Figure 1.10). This results in the potential to produce multiple distinct proteins from the same gene, adding greatly to the diversity of proteins encoded by the genome. Specific exons may correspond with particular functional domains of proteins, leading to the production of multiple proteins with diverse functions from the same gene. Some mRNAs are subject to RNA editing, wherein a specific base may be enzymatically modified. For example, the protein apolipoprotein B exists in two forms, a 48 kDa form made in the intestine and a 100 kDa form in the liver. Both forms are the product of the same gene. In the intestine the enzyme cytidine deaminase alters a C to a U at codon 2153, changing the codon from CAA (encoding glutamine) to UAA (a stop codon). This truncates the peptide, accounting for the 48 kDa form.
MicroRNA
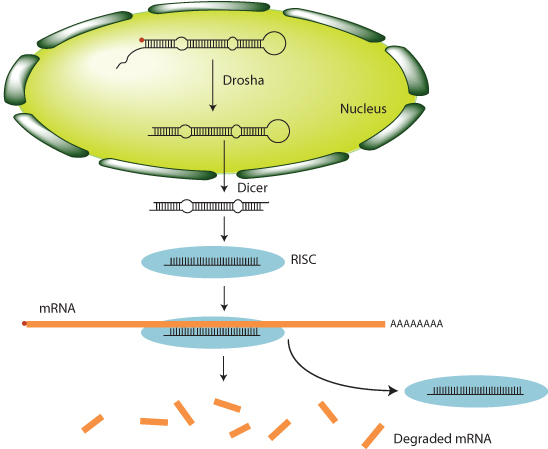
Gene regulation is not limited to control at the level of gene transcription. There is another level of posttranscriptional control that involves RNA molecules that do not encode protein, referred to as microRNAs (miRNAs) (Figure 1.11). Several hundred distinct miRNAs are encoded in the human genome. These are transcribed by RNA polymerase II, are capped, and have poly-A tails added. miRNAs form hairpin structures through base pairing. The enzyme Drosha trims the hairpins, which are then exported to the cytoplasm, where the enzyme Dicer further cleaves them. After further processing, single-stranded miRNA molecules associate with a protein complex called the RNA-induced silencing complex (RISC). The RISC then binds to mRNA molecules by base sequence complementarity with the miRNA. This leads to either cleavage and degradation of the mRNA, or reduced rate of translation and eventual degradation of the mRNA. The overall effect is for miRNA to reduce the quantity of protein produced from a transcribed gene. Any specific miRNA might bind to many different mRNAs, leading to coordinated reduction in gene product from a large number of genes.
A similar process also occurs when double-stranded RNA is introduced into the cell by viral infection. The Dicer enzyme cuts the invading RNA into shorter fragments, called small interfering RNA (siRNA), which attach to the RISC. The RISC then binds to additional double-stranded RNA molecules and causes their degradation. This is referred to as RNA interference, and represents a kind of immune system within the cell. The process has also been used experimentally, wherein siRNA molecules are introduced into the cell to selectively interfere with translation of targeted mRNAs.
Translation
The mature mRNA is exported to the cytoplasm for translation into protein. During translation, the mRNA sequence is read into the amino acid sequence of a protein (Figure 1.12). The translational machinery consists of a protein–RNA complex called the ribosome. Ribosomes consist of a complex of proteins and specialized rRNA. The eukaryotic ribosome is composed of two subunits, designated 60S and 40S (“S” is a measurement of density, the Svedberg unit, reflecting how the complexes were initially characterized by density gradient centrifugation). Each subunit includes proteins and an rRNA molecule. The 60S subunit includes a 28S rRNA, and the 40S subunit an 18S rRNA. Ribosomes can be free or associated with the endoplasmic reticulum (ER), also known as the rough ER.
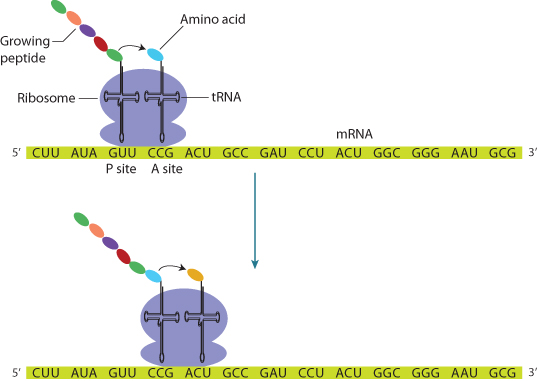
The mRNA sequence is read in triplets, called codons, beginning at the 5′ end of the mRNA, which is always AUG, encoding methionine (although this methionine residue is sometimes later cleaved off). Each codon corresponds with a particular complementary anticodon, which is part of another RNA molecule, tRNA. tRNA molecules bind specific amino acids defined by their anticodon sequence (Table 1.1). Protein translation therefore consists of binding a specific tRNA to the appropriate codon, which juxtaposes the next amino acid in the growing peptide, which is enzymatically linked by an amide bond to the peptide. The process ends when a stop codon is reached (UAA, UGA, or UAG). The peptide is then released from the ribosome for transport to the appropriate site within the cell, or for secretion from the cell. A leader peptide sequence may direct the protein to its final destination in the cell; this peptide is cleaved off upon arrival. Posttranslational modification, such as glycosylation, begins during the translation process and continues after translation is complete.
Table 1.1 The genetic code. A triplet codon is read from the left column, to the top row, to the full triplet in each box. Each codon corresponds with a specific amino acid, except for the three stop codons (labeled “Ter”). Most amino acids are encoded by more than one codon.
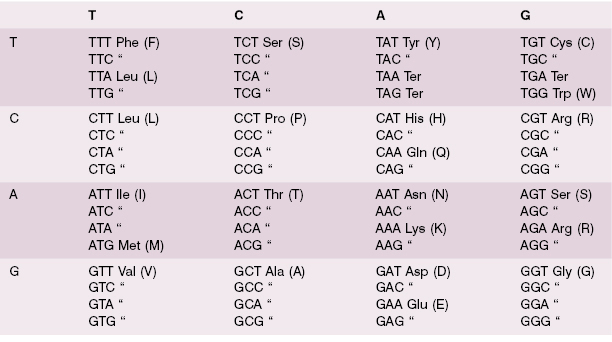
The process of translation consists of three phases, referred to as initiation, elongation, and termination. Initiation involves the binding of the first amino acyl tRNA, which always carries methionine, to the initiation codon, always AUG. A set of proteins, referred to as elongation factors, are involved in the process, which also requires adenosine triphosphate (ATP) and guanosine triphosphate (GTP). The ribosome binds to the mRNA at two successive codons. One is designated the P site and carries the growing peptide chain. The other is the next codon, designated the A site. Elongation involves the binding of the next amino acyl tRNA to its anticodon at the A site. This delivers the next amino acid in the peptide chain, which is attached to the growing peptide, with peptide bond formation catalyzed by peptidyl transferase. The ribosome then moves on to the next codon under the action of a translocase, with energy provided by GTP. When a stop codon is reached, a release factor protein–GTP complex binds and the peptidyl transferase adds an OH to the end of the peptide, which is then released from the ribosome under the influence of proteins called release factors.
Epigenetics
Individual genes may be reversibly activated or repressed, but there are some situations in which genes or sets of genes are permanently silenced. This occurs as a result of chemical modifications of DNA that do not change the base sequence, and also involves chemical changes in the associated histone proteins that result in compaction of the chromatin. Gene silencing is characteristic of one of the two copies of the X chromosome in females and on the maternal or paternal copy of imprinted genes. It also can occur on other genes in specific tissues and may be subject to environmental influences.
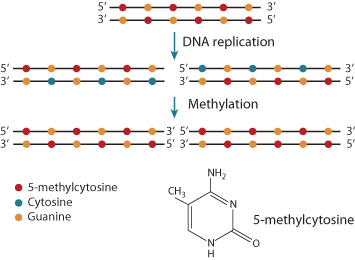
At the DNA level, gene silencing is accompanied by methylation of cytosine bases to 5-methylcytosine (Figure 1.13). This occurs in regions where cytosine is following by guanine (5′–CpG–3′) near the promoter, sites referred to as CpG islands. Methylated sites bind protein complexes that remove acetyl groups from histones, leading to transcriptional repression. The silencing is continued from cell generation to generation because the enzymes responsible for methylation recognize the 5-methylcytosine on the parental strand of DNA and methylate the cytosine on the newly synthesized daughter strand.
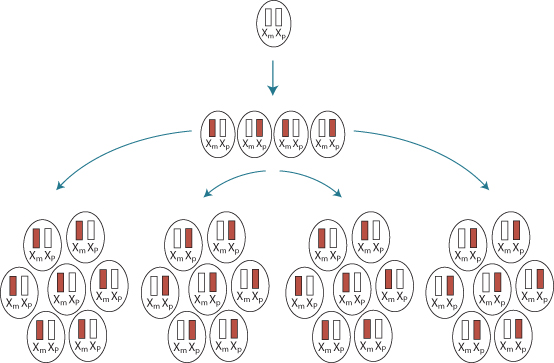
X-chromosome inactivation provides a mechanism for equalization of gene dosage on the X chromosome in males, who have one X, and females, who have two. Most genes on one of the two X chromosomes in each cell of a female are permanently inactivated early in development (Figure 1.14). The particular X inactivated in any cell is determined at random, so in approximately 50% of cells one X is inactivated and in the other 50% the other X is inactivated. Regions of homology between the X and Y at the two ends of the X escape inactivation. These are referred to as pseudoautosomal regions. The inactive X remains condensed through most of the cell cycle, and can be visualized as a densely staining body during interphase, called the Barr body.
Initiation of inactivation is controlled from a region called the X inactivation center (Xic). A gene within this region, known as Xist, is expressed on one of the two X chromosomes early in development. Xist encodes a 25 kb RNA that is not translated into protein, but binds to sites along the X to be inactivated. Subsequently, CpG islands on this chromosome are methylated and histones are deacetylated.
Genomic imprinting involves the silencing of either the maternal or paternal copy of a gene during early development (Figure 1.15