
Table of Contents
Related Titles
Title Page
Copyright
Preface
List of Contributors
Chapter 1: Stereoselective Acetate Aldol Reactions
1.1 Introduction
1.2 Mukaiyama Aldol Reaction
1.3 Metal Enolates
1.4 Conclusions
References
Chapter 2: The Vinylogous Mukaiyama Aldol Reaction in Natural Product Synthesis
2.1 Introduction
2.2 Aldehyde-Derived Silyl Dienol Ethers
2.3 Ester-Derived Silyl Dienol Ethers
2.4 Amide-Derived Silyl Dienol Ethers – Vinylketene Silyl N,O-Acetals
2.5 Acyclic Acetoacetate-Derived Silyl Dienolates – Chan's Diene
2.6 Cyclic Acetoacetate-Derived Dienolates
2.7 Furan-Derived Silyloxy Dienes
2.8 Pyrrole-Based 2-Silyloxy Dienes
2.9 Comparison with Other Methods
References
Chapter 3: Organocatalyzed Aldol Reactions
3.1 Introduction
3.2 Proline as Organocatalyst
3.3 Proline Derivatives as Organocatalysts
3.4 Conclusions and Outlook
References
Chapter 4: Supersilyl Protective Groups in Aldol Reactions
4.1 Introduction
4.2 Aldol Addition with Acetaldehyde-Derived Super Silyl Enol Ether (1)
4.3 α-Substituted Silyl Enol Ethers Derived from Aldehydes
4.4 Aldol Addition to Chiral Aldehydes
4.5 One-Pot Sequential Aldol Reactions
4.6 Sequential Aldol–Aldol Reactions of Acetaldehyde
4.7 Double Aldol Reactions with α-Substituted Silyl Enol Ethers
4.8 Stereochemical Considerations
4.9 Aldol Reactions of β-Supersiloxy Methyl Ketones
4.10 Total Synthesis of Natural Products Using Supersilyl Aldol Reactions
4.11 Conclusion and Outlook
References
Chapter 5: Asymmetric Induction in Aldol Additions
5.1 Introduction
5.2 Asymmetric Induction Using Chiral Ketones
5.3 Asymmetric Induction Using Chiral Aldehydes
5.4 Asymmetric Induction in the Aldol Addition of Chiral Enolates to Chiral Aldehydes
References
Chapter 6: Polypropionate Synthesis Via Substrate-Controlled Stereoselective Aldol Couplings of Chiral Fragments
6.1 Introduction
6.2 Principles of Stereoselective Aldol Reactions
6.3 Stereoselective Aldol Coupling of Chiral Reactants
6.4 2-Alkoxyalkyl Ethyl Ketones: 2-Desmethyl Polypropionate Equivalents
6.5 Conclusions
References
Chapter 7: Application of Oxazolidinethiones and Thiazolidinethiones in Aldol Additions
7.1 Introduction
7.2 Preparation of Oxazolidinethione and Thiazolidinethione Chiral Auxiliaries
7.3 Acylation of Oxazolidinethione and Thiazolidinethione Chiral Auxiliaries
7.4 Propionate Aldol Additions
7.5 Acetate Aldol Additions
7.6 Glycolate Aldol Additions
References
Chapter 8: Enzyme-Catalyzed Aldol Additions
8.1 Introduction
8.2 Pyruvate Aldolases
8.3 N-Acetylneuraminic Acid Aldolase (NeuA)
8.4 Dihydroxyacetone Phosphate (DHAP) Aldolases
8.5 D-Fructose-6-Phosphate Aldolase and Transaldolase B Phe178Tyr: FSA-Like Aldolases
8.6 2-Deoxy-D-Ribose-5-Phosphate Aldolase (RibA or DERA; EC 4.1.2.4)
8.7 Glycine/Alanine Aldolases
8.8 Aldol Reactions Catalyzed by Non aldolases
8.9 Conclusions and Perspectives
References
Index
Related Titles
Majumdar, K. C., Chattopadhyay, S. K. (eds.)
Heterocycles in Natural Product Synthesis
2011
ISBN: 978-3-527-32706-5
Poupon, E., Nay, B. (eds.)
Biomimetic Organic Synthesis
2011
ISBN: 978-3-527-32580-1
Nicolaou, K. C., Chen, J. S.
Classics in Total Synthesis III
Further Targets, Strategies, Methods
2011
ISBN: 978-3-527-32958-8
Yang, J
Six-Membered Transition States in Organic Synthesis
2010
ISBN: 978-0-470-65258-9
Joule, J. A., Mills, K.
Heterocyclic Chemistry
2010
ISBN: 978-0-470-65258-9
Zabicky, J. (ed.)
The Chemistry of Metal Enolates
2009
ISBN: 978-0-470-06168-8
Carreira, E. M., Kvaerno, L.
Classics in Stereoselective Synthesis
2009
ISBN: 978-3-527-32452-1

The Editor
Prof. Dr. Rainer Mahrwald
Humboldt-Universität Berlin
Institut für Chemie
Brook-Taylor-Str. 2
12489
Berlin
All books published by Wiley-VCH are carefully produced. Nevertheless, authors, editors, and publisher do not warrant the information contained in these books, including this book, to be free of errors. Readers are advised to keep in mind that statements, data, illustrations, procedural details or other items may inadvertently be inaccurate.
Library of Congress Card No.: applied for
British Library Cataloguing-in-Publication Data
A catalogue record for this book is available from the British Library.
Bibliographic information published by the Deutsche Nationalbibliothek
The Deutsche Nationalbibliothek lists this publication in the Deutsche Nationalbibliografie; detailed bibliographic data are available on the Internet at <http://dnb.d-nb.de>.
© 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Boschstr. 12, 69469 Weinheim, Germany
All rights reserved (including those of translation into other languages). No part of this book may be reproduced in any form — by photoprinting, microfilm, or any other means — nor transmitted or translated into a machine language without written permission from the publishers. Registered names, trademarks, etc. used in this book, even when not specifically marked as such, are not to be considered unprotected by law.
Print ISBN: 978-3-527-33205-2
ePDF ISBN: 978-3-527-65674-5
ePub ISBN: 978-3-527-65673-8
oBook ISBN: 978-3-527-65671-4
mobi ISBN: 978-3-527-65672-1
Cover Design Adam-Design, Weinheima
Typesetting Laserwords Private Limited, Chennai, India
Preface
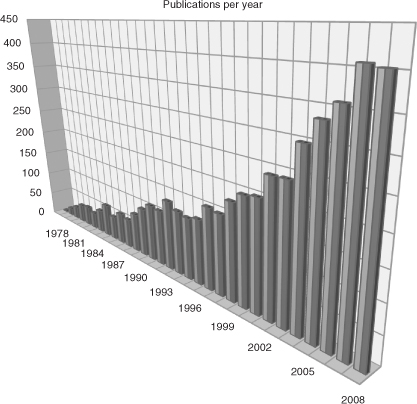
Stereoselectivity is one of the most important aspects for natural product chemists. Following the increasing possibility of detection and assignment of stereogenic centers, a tremendous increase in stereoselective methods of organic reactions, particularly aldol reactions, has been noticed. In the beginning of this development, only sporadic examples of stereoselective aldol reactions were described, mostly in the context of total syntheses of natural products. An outstanding early example is the R. B. Woodward's proline-catalyzed aldol addition in the total synthesis of erythronolide A at the Harvard University in 1981. In the following three decades, a vast arsenal of stereoselective aldol additions has been developed (see Figure).
This book provides a comprehensive review of modern aldol reactions, especially in the aspect of how to achieve high stereoselectivity – diastereoselectivity as well as enantioselectivity. Stereoselection is discussed under several different aspects. One aspect is the deployment of different substrates – acetate or propionate aldol reactions. Another aspect is the mode of action including metal enolate chemistry, Lewis acid as well as Lewis base catalysis, enzymatic catalysis, and organocatalysis. There are some overlappings of these aspects in the chapters covering the cross-cutting themes of vinyloguos Mukaiyama reaction or asymmetric inductions (e.g., compare Scheme 1.50 with Scheme 2.59) or total synthesis of dolastatin 19 – (compare Scheme 1.82 with Scheme 5.8). These overlappings, however, are intentional in order to give a comprehensive insight into the techniques for installing required configurations during aldol reactions. The utility of the corresponding methods is shown in the context of total syntheses of natural products. All chapters are thoroughly well written by experts in the respective fields.
It is my pleasure to express profound gratitude to the 15 authors for their huge endeavor to organize and summarize this vast amount of material. It has been a great pleasure for me to work with this team of authors at all times. Finally, my special thanks go to Elke Maase and Bernadette Gmeiner at WILEY for their fine work in making this book a reality.
Berlin, Autumn 2012
Rainer Mahrwald
List of Contributors
Chapter 1
Stereoselective Acetate Aldol Reactions
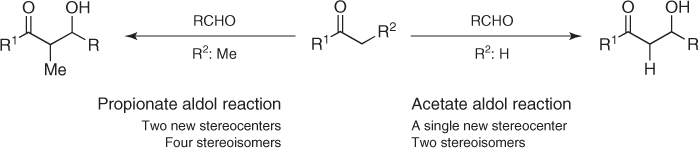
The stereochemical control of aldol reactions from unsubstituted enol- or enolatelike species, what are known as acetate aldol reactions, has been a matter of concern for nearly 30 years [1, 2]. Indeed, pioneering studies soon recognized that the asymmetric installation of a single stereocenter in such aldol reactions was much more demanding than the simultaneous construction of two new stereocenters in the related propionate counterparts (Scheme 1.1) [3]. This challenge, together with the ubiquitous presence of chiral β-hydroxy α-unsubstituted oxygenated structures in natural products, has motivated the development of new concepts and strategies and a large number of highly stereoselective methodologies. These involve Lewis-acid-mediated additions of enolsilane derivatives of carbonyl compounds to aldehydes (Mukaiyama aldol variant) [4, 5], a plethora of transformations that take advantage of the reactivity of boron, titanium(IV), and tin(II) enolates (metal enolates) [6], and some insightful organocatalytic approaches [7]. In spite of these accomplishments, the quest for more powerful and selective methodologies and a better understanding of their intricate mechanisms is an active area of research. Herein, we describe the most significant achievements in the field of stereoselective acetate aldol reactions based on the Lewis-acid-mediated addition of enolsilanes and metal enolates to aldehydes, with particular attention to their application to the asymmetric synthesis of natural products. Recent advances in parallel organocatalytic procedures are not discussed.
Scheme 1.1 Aldol reactions.
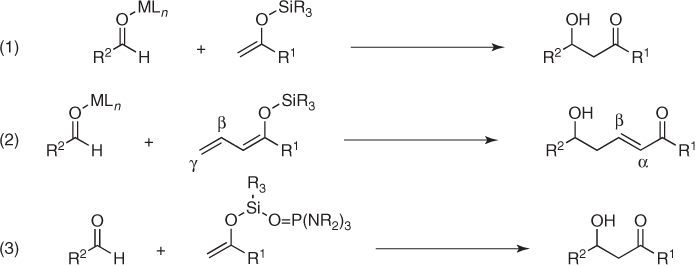
With some significant exceptions, enolsilanes are unreactive toward aldehydes.1 This lack of reactivity can be overcome by increasing the electrophilic character of aldehydes or the nucleophilicity of enolsilanes. The former option is achieved by coordination of Lewis acids (MLn) to the carbonyl group, which enhances the electrophilicity of the C—O bond and facilitates the attack of enolsilanes. This represents the canonical Mukaiyama aldol variant ((1) in Scheme 1.2) [4, 5]. It also covers vinylogous aldol transformations, which involve the reactions of γ-unsubstituted β, γ-conjugated enolsilanes ((2) in Scheme 1.2) [8]. In turn, the latter option takes advantage of the activation of the nucleophilic character of enolsilanes by binding of Lewis bases such as phosphoramides (O—P(NR2)3) to the silicon atom ((3) in Scheme 1.2) [9].
Scheme 1.2 Mukaiyama aldol variants.
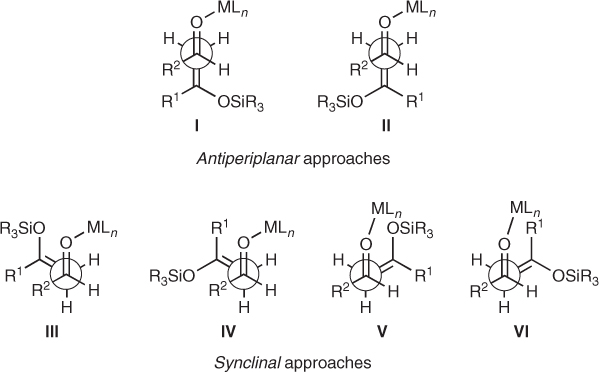
Early mechanistic analyses suggested that Lewis-acid-mediated aldol reactions represented in Scheme 1.2 proceeded through open transition states [4, 5, 10]. This model assumes a transoid geometry for the Lewis-acid-aldehyde complex, which the enolsilane attacks following antiperiplanar or synclinal approaches, as represented in Scheme 1.3. Antiperiplanar transition states I and II are usually more favorable because of the minimization of dipolar interactions, the steric interactions between the enolsilane (R1 or R3SiO groups) and the aldehyde (R2 group) being the main source of instability. Similar steric interactions arise in synclinal transition states III and IV, whereas V and VI are characterized by a destabilizing interaction between the enolsilane and the Lewis acid coordinated to the carbonyl oxygen. Then, steric and stereoelectronic interactions determine the relative stability of I–VI and the capacity to differentiate one from the other faces of the carbonyl bond.
Scheme 1.3 Open transition states for Mukaiyama aldol reactions.
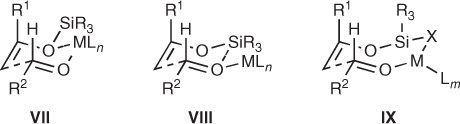
Despite the importance and utility of this paradigm, it is probably an oversimplified model because it ignores the fate of the silyl group. In this respect, some models take into account the silicon moiety and suggest cyclic transition states VII–IX, as represented in Scheme 1.4. Importantly, the role of the silyl group is not limited to influencing the nature of the transition state, because the silicon transfer from the enolsilane to the β-alkoxy position may be a key step in the overall mechanism and becomes crucial to the turnover necessary for nonstoichiometric transformations [11].
Scheme 1.4 Cyclic transition states for Mukaiyama aldol reactions.
Irrespective of the mechanistic pathway, the asymmetric induction achieved by these Lewis-acid-mediated aldol reactions depends on chiral elements on the enolsilane (the nucleophilic partner), the aldehyde (the electrophilic partner), or the Lewis acid (the activating element), so they must all cooperate to provide the appropriate face differentiation of the carbonyl bond in order to control the configuration of the new stereocenter. The influence of these elements is discussed in the following sections.
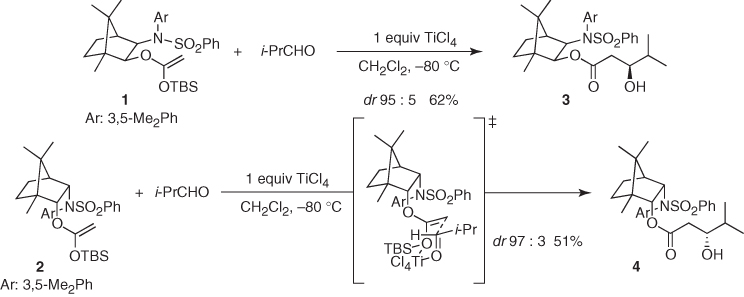
In the context of emergence of chiral auxiliaries as powerful platforms to achieve asymmetric transformations, Helmchen reported highly diastereoselective aldol reactions of chiral auxiliary-based silyl ketene acetals (1) and (2) [12, 13]. As shown in Scheme 1.5, TiCl4-mediated additions of 1 and 2 to isobutyraldehyde afforded aldol adducts 3 and 4 in good yields and excellent diastereomeric ratios, presumably through a chairlike transition state in which the titanium atom is simultaneously coordinated to the carbonyl and the OTBS group. In turn, these adducts can be converted into the corresponding β-hydroxy acids in quantitative yield by simple treatment with KOH in methanol.
Scheme 1.5 Chiral auxiliary-based Mukaiyama aldol reactions.
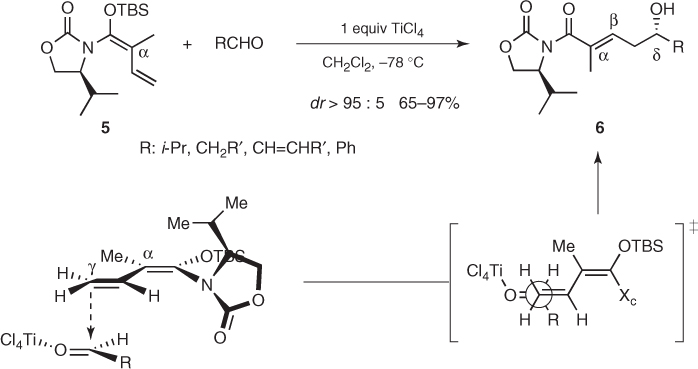
This approach was quickly surpassed by alternative methodologies based on chiral aldehydes or Lewis acids and bases (Sections 1.2.4–1.2.6). Nevertheless, new findings restored the interest in this sort of transformations a few years ago. Indeed, Kobayashi described highly stereoselective vinylogous Mukaiyama aldol reactions using silyl vinyl ketene N,O-acetals prepared from valine-derived 1,3-oxazolidin-2-ones [14]. As represented in Scheme 1.6, TiCl4-mediated additions of α-methyl acetal (5) to aliphatic, α, β-unsaturated, and aromatic aldehydes afforded δ-hydroxy-α-methyl-α, β-unsaturated imides (6) in excellent yields and diastereomeric ratios. Such outstanding remote asymmetric induction was believed to arise from a conformation in which the chiral heterocycle is almost perpendicular to the dienol plane and the isopropyl group overhangs the upper face of the dienol moiety. Then, the aldehyde approaches from the less hindered face through an open transition state in which the α-methyl group appears to be essential to achieve the observed high stereocontrol. Finally, the chiral auxiliary can be removed by well-known methodologies used for Evans auxiliaries.
Scheme 1.6 Chiral auxiliary-based vinylogous Mukaiyama aldol reactions.
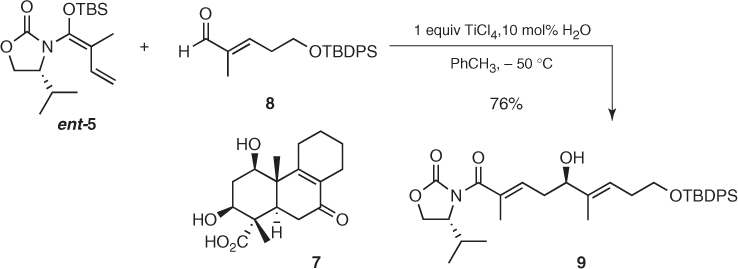
This methodology was used for the construction of the AB ring of fomitellic acids (7) (Scheme 1.7) [15]. Initially, application of the standard conditions to ent-5 and enal (8) provided the desired aldol (9), but the reaction was slow and hard to reproduce. Then, a thorough study of this particular reaction uncovered significant rate enhancements by adding catalytic amounts of water in toluene, which permitted to obtain aldol (9) in 76% yield as a single diastereomer in a straightforward and consistent way [16]. The origin of this catalytic effect remains unclear, but it has proved to be general and has been successfully applied to other aldehydes [17].
Scheme 1.7 Synthesis of the central ring of fomitellic acids.
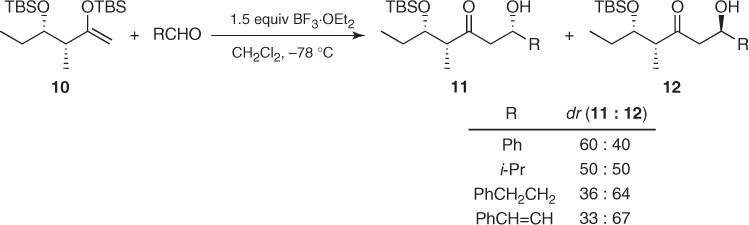
There are no systematic studies on the asymmetric induction imparted by chiral methyl ketones. However, most of the examples reported so far suggest that substrate-controlled Mukaiyama aldol reactions based on chiral methyl ketones are poorly stereoselective. This lack of stereocontrol is well illustrated by the aldol reaction of chiral silyl enol ether (10), in which the major diastereomer, 11 or 12, depends on the achiral aldehyde (Scheme 1.8) [18].
Scheme 1.8 Asymmetric induction imparted by a silyl enol ether from a chiral methyl ketone.
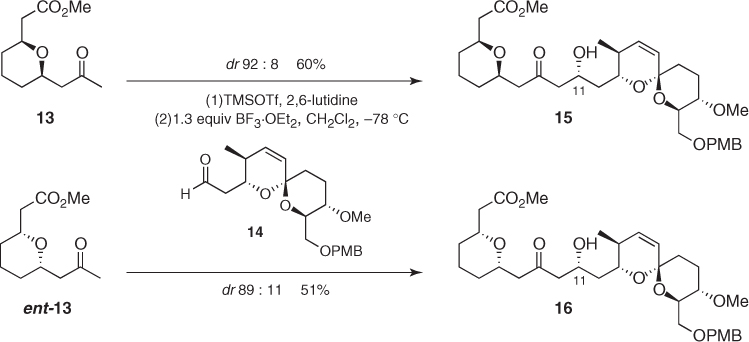
In a more complex framework, De Brabander also reported that silyl enol ethers from enantiomeric methyl ketones (13) and (ent-13) underwent additions to chiral aldehyde (14) to afford the corresponding aldol adducts 15 and 16 in similar yields (Scheme 1.9) [19]. Considering that the new C11-stereocenters possess the same configuration in both adducts and that the diastereoselectivity is comparable for both processes, it can be concluded that the asymmetric induction provided by the aldehyde is much more important than that provided by the ketone.
Scheme 1.9 Chiral methyl ketones in stereoselective Mukaiyama aldol reactions.
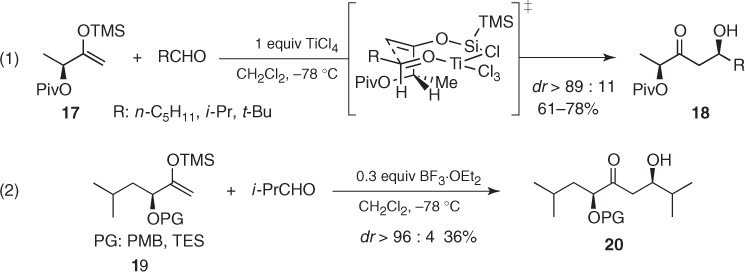
Lactate-derived and other α-hydroxy methyl ketones are exceptions to this trend. Thus, Trost found that TiCl4-mediated aldol reactions of pivaloyl-protected silyl enol ether (17) afforded β-hydroxy ketones (18) in high yields and diastereomeric ratios up to 98 : 2 [20]. This was assumed to be achieved through an eight-membered cyclic transition state in which the titanium is simultaneously bound to the aldehyde and the enolsilane ((1) in Scheme 1.10). Importantly, dipole–dipole interactions are understood to favor the antiperiplanar arrangement of the C–OPiv and the C–OSi bonds and impel the aldehyde toward the less hindered face of the enolsilane. Moreover, Kalesse reported that parallel BF3-catalyzed additions of silyl enol ethers (19) to isobutyraldehyde afforded the corresponding aldols (20) with excellent diastereoselectivity but in low yield ((2) in Scheme 1.10) [21]. The origin of such remarkable stereocontrol is unclear.
Scheme 1.10 Asymmetric induction imparted by chiral α-hydroxy methyl ketones in Mukaiyama aldol reactions.
At this point, it is worth mentioning that Yamamoto has also reported highly diastereoselective Mukaiyama aldol reactions based on chiral β-tris(trimethylsilyl)silyloxy methyl ketones containing a single stereocenter at the β-position [22]. This chemistry is discussed in connection with parallel methodologies (Sections 1.2.4 and 1.3.6).
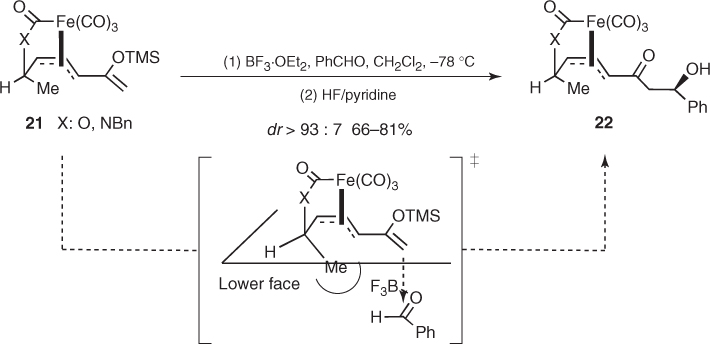
Finally, Ley reported that reactions of silyl enol ethers from chiral π-allyltricarbonyliron lactone or lactam complexes proceeded with a significant remote stereocontrol [23]. This is illustrated by the BF3-mediated addition of complexes 21 to benzaldehyde furnishing β-hydroxy ketones (22) with excellent diastereomeric ratios (Scheme 1.11). The remarkable 1,7-induction provided by these substrates is due to the chiral environment created on the lower face of the silyl enol ether (the upper face is blocked by the tricarbonyliron moiety) by the endo-oriented methyl substituent at the sp3-stereocenter. Then, the incoming activated aldehyde approaches in a synclinal arrangement in which unfavorable steric interactions are minimized.
Scheme 1.11 Asymmetric induction imparted by chiral π-allyltricarbonyl iron complexes in Mukaiyama aldol reactions.
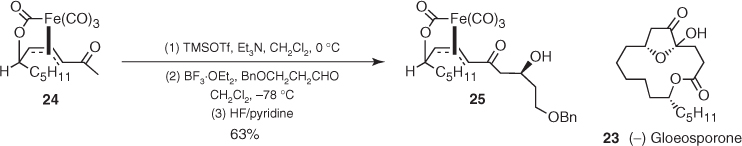
As the iron lactone and lactam (22) can be easily decomplexed to afford a rich array of stereodefined derivatives, this reaction may represent a powerful tool to the rapid construction of highly functionalized systems under remote stereocontrol. For instance, total synthesis of (−)-gloeosporone (23) commenced with the addition of silyl enol ether from methyl ketone (24) to benzyloxypropanal, which afforded aldol (25) as a single diastereomer in a 63% yield (Scheme 1.12) [24].
Scheme 1.12 Synthesis of (−)-gloeosporone.
The asymmetric induction of chiral aldehydes in Mukaiyama aldol reactions is much more important and has stimulated the formulation of increasingly more refined models to predict the π-facial selectivity in nucleophilic additions to the carbonyl bond [25]. Therefore, the influence of α- and β-substituents has received particular attention and is described in detail in the following sections.
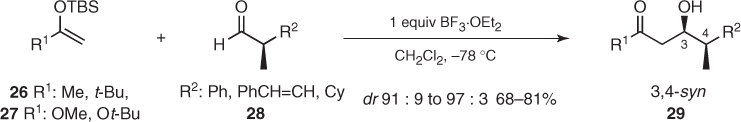
Pioneering studies on acyclic stereoselection established that Mukaiyama acetate aldol additions of enolsilane derivatives (26) and (27) to chiral α-methyl aldehydes (28) proceeded with high diastereofacial selectivity to favor 3,4-syn aldol adducts (29) (Scheme 1.13) [26].
Scheme 1.13 Asymmetric induction imparted by chiral α-methyl aldehydes in Mukaiyama aldol reactions.
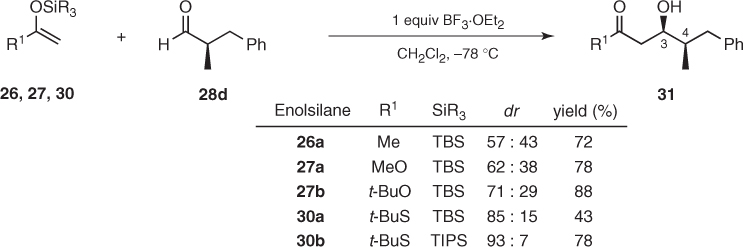
As expected, the 1,2-asymmetric induction of such aldehydes was eroded when R2 was sterically similar to the α-methyl. The challenge posed by these transformations can be met by using more bulky nucleophiles, as has been observed in the aldol additions of enolsilanes (26), (27), and (30) to 2-methyl-3-phenylpropanal (Scheme 1.14). The stereochemical outcome of these reactions shows that enhancement of the steric hindrance of R1 and SiR3 groups gives the corresponding 3,4-syn aldols (31) in higher diastereomeric ratios [26, 27]. A parallel improvement can also be attained by employing more bulky Lewis acids, but steric influences must be analyzed carefully because some combinations of bulky nucleophiles and Lewis acids do not provide the expected results [27].
Scheme 1.14 Mukaiyama aldol additions to (R) 2-methyl-3-phenylpropanal.
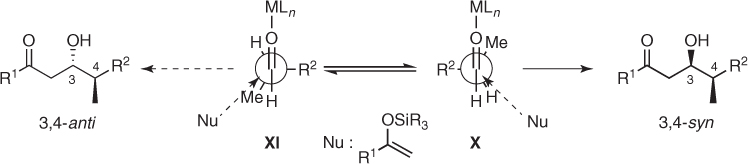
The Felkin–Anh model [25, 28] is usually invoked to account for the asymmetric induction observed in the Mukaiyama aldol additions to these chiral α-methyl aldehydes. Thus, once the methyl group has been identified as the medium size group, the major 3,4-syn diastereomer is obtained by bringing the enolsilane close to the face of the C—O bond in which the steric interactions between the nucleophile and the α-substituent (H vs Me) are weaker (X in Scheme 1.15).
Scheme 1.15 The Felkin–Anh model for Mukaiyama aldol additions to chiral α-methyl aldehydes.
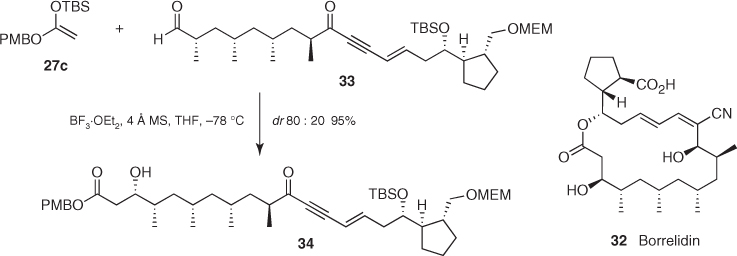
The total synthesis of borrelidin (32) reported by Theodorakis contains a good example of stereocontrol based on the asymmetric induction imparted by such chiral aldehydes [29]. As represented in Scheme 1.16, the Mukaiyama aldol addition of silyl ketene acetal (27c) to α-methyl aldehyde (33) produced the desired ester (34) as a 80 : 20 mixture of diastereomers in a 95% combined yield, which demonstrates the π-facial selectivity provided by the α-stereocenter of aldehydes of this kind [30].
Scheme 1.16 Synthesis of borrelidin.
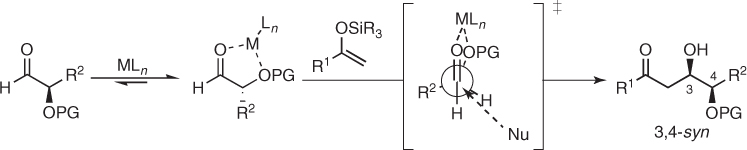
The substitution of the methyl group by a heteroatom affects these transformations dramatically. Indeed, a tenet in asymmetric synthesis states that nucleophilic additions to chiral aldehydes bearing an α-heteroatom attain outstanding levels of stereocontrol provided that the reaction is carried out under conditions in which chelate organization is favored. In this context, the Cram model [25, 31] accounts for the stereochemical outcome of chelate-controlled Mukaiyama aldol reactions. According to this model, the appropriate choice of the Lewis acid and the protecting group of α-hydroxy aldehydes permits the formation of stable five-membered chelated complexes and gives the corresponding 3,4-syn aldol adducts in a highly diastereoselective manner, presumably through an open transition state in which the nucleophile approaches the less hindered face of the chelated carbonyl group (Scheme 1.17).
Scheme 1.17 Cram model for Mukaiyama aldol additions to chiral α-hydroxy aldehydes.
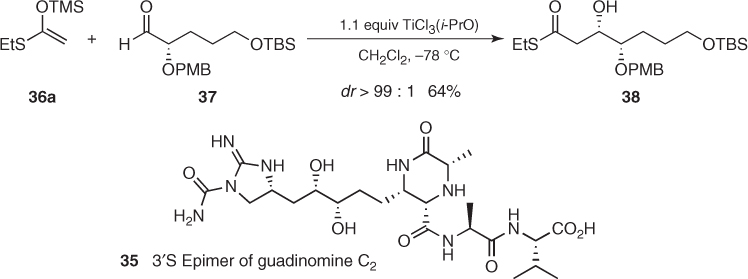
This highly reliable and powerful element of stereocontrol has been widely exploited in the synthesis of natural products. For instance, Sunazuka and Omura used a chelate-controlled Mukaiyama aldol reaction for the total synthesis of an epimer of guadinomine C2 (35). As shown in Scheme 1.18, addition of silyl ketene S,O-acetal (36a) to chiral α-OPMB aldehyde (37) in the presence of 1.1 equiv of TiCl3(i-PrO) gave 3,4-syn aldol (38) with exceptional diastereoselectivity in 64% yield [32].
Scheme 1.18 Synthesis of (3′S) epimer of guadinomine C2.
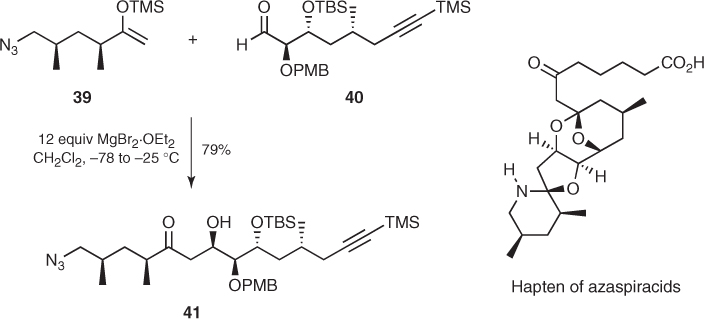
Moreover, Forsyth reported that the addition of mild Lewis acid MgBr2 · OEt2 to a mixture of chiral silyl enol ether (39) and α-OPMB aldehyde (40) triggered a smooth aldol reaction that furnished 3,4-syn aldol (41) as a single diastereomer in 79% yield, which was further elaborated to a hapten for azaspiracids (Scheme 1.19) [33]. A very similar transformation was also reported by Evans [34, 35].
Scheme 1.19 Synthesis of a hapten for azaspiracids.
In the absence of chelated intermediates, nucleophilic additions to chiral aldehydes possessing an α-heteroatom are currently explained by the polar Felkin–Anh [36] and Cornforth models [37], which apply to conformations XII–XV arising from rotation about the C1–C2 bond of the aldehyde (Scheme 1.20) [25]. The polar Felkin–Anh model is based on the premise that staggered transition states positioning the C–X bond perpendicularly to the carbonyl bond are preferred ((1) in Scheme 1.20). In turn, the Cornforth model embraces the assumption that electrostatic effects are instrumental in dictating a nearly antiparallel relationship between the carbonyl and the C–X bond ((2) in Scheme 1.20). Then, the stereochemical outcome of these additions depends on the steric interactions between the nucleophile and the remaining α-substituents in alternative transition states. Application of both models to Mukaiyama aldol reactions predicts the preferential formation of 3,4-anti aldol adducts.
Scheme 1.20 Models for Mukaiyama aldol additions to chiral aldehydes possessing an α-heteroatom.
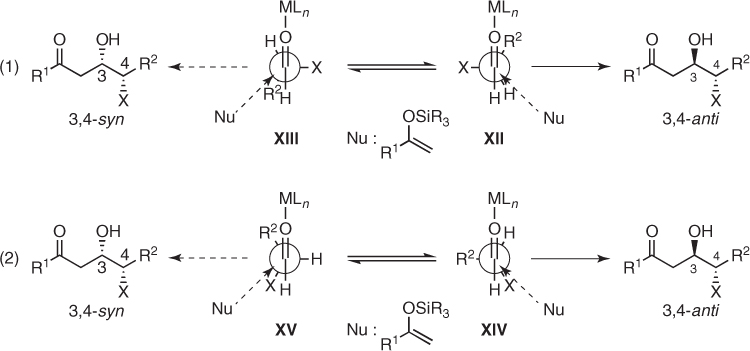
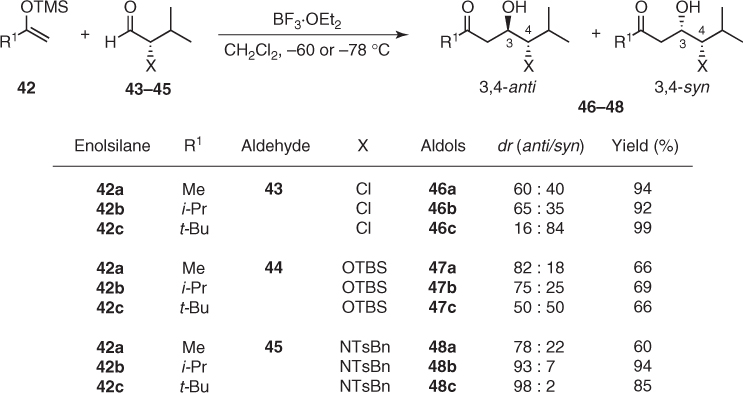
Most of the aldol reactions involving such aldehydes proceed in accordance with these expectations, but systematic studies on the addition of enolsilanes (42) to α-chloro, α-hydroxy, and α-amino aldehydes (43–45) revealed that their diastereoselectivity is dependent significantly on the α-heteroatom and the steric bulk of nucleophiles (Scheme 1.21) [38–40]. Thus, additions of acetone-derived enolsilane (42a) to aldehydes (43) and (44) possessing an electronegative α-heteroatom such as chlorine or oxygen afforded the corresponding 3,4-anti aldols (46a) and (47a) (dr 60 : 40 and 82 : 18, respectively), whereas more sterically hindered pinacolone-derived enolsilane (42c) gave under the same conditions 3,4-syn aldol (46c) or equimolar mixtures of 3,4-anti and syn diastereomers (47c). In turn, N-benzyl-N-tosyl protected valinal (45) always furnished 3,4-anti aldols (48) in modest to excellent diastereomeric ratios (Scheme 1.21).
Scheme 1.21 Mukaiyama aldol additions to chiral aldehydes possessing an α-heteroatom.
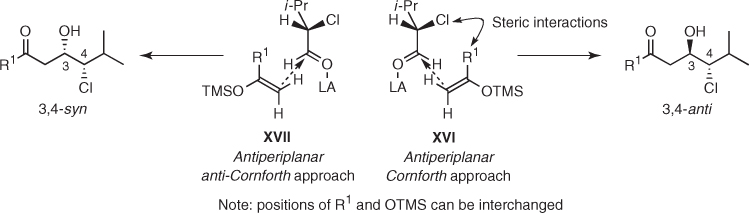
In view of these and a few related studies on the influence of bulky Lewis acids, Somfai proposed that N-protected valinal (45) prefers the polar Felkin–Anh manifold, which is not seriously affected by the size of the nucleophile ((1) in Scheme 1.20) [39]. In turn, Mukaiyama aldol additions to α-chloro aldehyde (43) would proceed essentially through antiperiplanar Cornforth transition state XVI provided that R1 is small enough to avoid serious steric interactions with chlorine (Scheme 1.22). Facing such deleterious interactions, sterically hindered silyl enol ethers would react through antiperiplanar anti-Cornforth transition state XVII. A similar rationale could be applied to α-silyloxy aldehyde (44).
Scheme 1.22 Cornforth model for Mukaiyama aldol additions to (S) 2-chloro-3-methylbutanal.
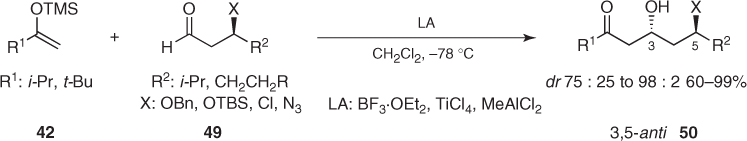
Mukaiyama aldol additions to chiral α-unsubstituted aldehydes bearing a β-heteroatom usually proceed with significant levels of 1,3-anti asymmetric induction [41]. As illustrated in Scheme 1.23, most of the aldol reactions of silyl enol ethers (42) with β-hydroxy, β-chloro, and β-azido aldehydes (49) produce the corresponding 3,5-anti aldols (50) in high diastereomeric ratios regardless of the chelating ability of the Lewis acid and the hydroxyl protecting group [42–44].
Scheme 1.23 Asymmetric induction imparted by chiral α-unsubstituted aldehydes bearing a β-heteroatom in Mukaiyama aldol reactions.
The stereochemical outcome of these reactions may not be readily interpreted because both open-chain and chelation control lead to the same 3,5-anti diastereomer (50). Thus, Evans proposed, after a systematic and insightful study, a revised induction polar model and a chelate-controlled model to account for the high 1,3-asymmetric induction provided by such chiral aldehydes [43]. The former is an open-chain model ((1) in Scheme 1.24) derived from several assumptions common to the Felkin–Anh analysis for 1,2-asymmetric induction. First, it is assumed that torsional effects dictate that aldehyde transition state conformations adopt a staggered relationship between the forming bond and the aldehyde α-substituents. Second, it is also assumed that the principal diastereomer arises from the reactant-like transition state wherein the β-stereocenter is oriented anti, rather than gauche, to the forming bond because this geometry reduces nonbonded interactions between the aldehyde α-substituents and the incoming nucleophile. In turn, the chelate-controlled model applies to suitable β-hydroxy-protected aldehydes able to form stable six-membered chelates, which can subsequently react through transition states involving either boat or half-chair conformations ((2) in Scheme 1.24).
Scheme 1.24 Evans models for Mukaiyama aldol additions to chiral α-unsubstituted aldehydes bearing a β-heteroatom.
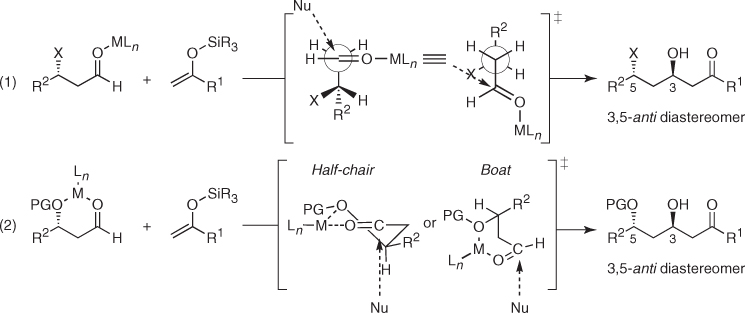
Regardless of mechanistic considerations, the predictable and high stereocontrol provided by these aldehydes has been used in many total syntheses. Evans reported in the total synthesis of bryostatin 2 (51) that aldol coupling of bis(trimethylsilyl)dienol ether (52a) with β-OPMB aldehyde (53) ((1) in Scheme 1.25) was only modestly stereoselective when it was carried out in the presence of MgBr2 · OEt2 or BF3 · OEt2, whereas strong Lewis acid (TiCl4, SnCl4) did not effect a clean transformation. Alternatively, mild TiCl2(i-PrO)2 in CH2Cl2 at − 78 °C delivered aldol (54) in a high-yielding and stereoselective manner (dr 86 : 14, 93%). Importantly, the diastereoselectivity was improved by using toluene (dr 94 : 6, 83%), a result that is consistent with the operation of electrostatic effects as the stereochemical control element [45]. In turn, Panek took advantage of the 1,3-asymmetric induction of β-alkoxy aldehydes in a challenging Mukaiyama aldol reaction to assemble an advanced intermediate in the total synthesis of leucascandrolide A (55) [46]. Indeed, the coupling of chiral silyl enol ether (56) and aldehyde (57) ((2) in Scheme 1.25) afforded the desired anti aldol (58) in 81% yield and excellent diastereomeric ratio (dr > 94 : 6). This, presumably occurred through an open transition state in which the β-stereocenter of the aldehyde determines the approach of the nucleophile to the carbonyl in accordance with the revised polar model described in (1) of Scheme 1.24. Finally, Nelson also used this reactivity in the total synthesis of the apoptolidine C aglycone (59) [47]. In this case, silyl enol ether (60) participated in a highly stereoselective addition to chiral aldehyde (61) ((3) in Scheme 1.25), affording aldol (62) as a single diastereomer in 71% yield. A matched pairing of aldehyde and enolsilane facial biases acting in the transition state shown in Scheme 1.25 was invoked to explain such outstanding stereoselectivity [48].
Scheme 1.25 Use of 1,3-asymmetric induction imparted by chiral β-hydroxy aldehydes in Mukaiyama aldol reactions in the synthesis of natural products.
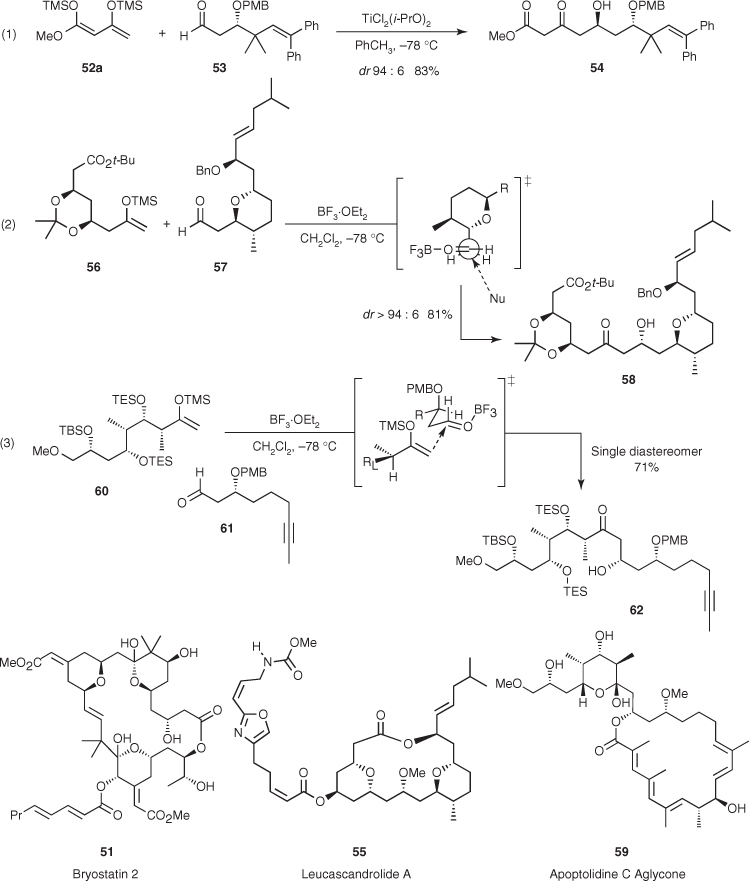
The aforementioned transformations show that the 1,3-anti asymmetric induction of protected β-hydroxy aldehydes can be very successful, but this control element should be used with caution because the diastereoselectivity drops when the steric bulk of the protecting group increases [49, 50]. Taking advantage of this trend, Yamamoto reported that the extremely bulky tris(trimethylsilyl)silyl group (TTMSS, (Me3Si)3Si), also known in the literature as the hypersilyl or super silyl group, confers to β-OTTMSS aldehydes an outstanding 1,3-syn asymmetric induction [51]. This silicon-protecting group is also remarkable because it permits acetaldehyde derived TTMSS enol ether to participate in highly diastereoselective Mukaiyama aldol additions to a large variety of aldehydes.2 Moreover, these additions can incorporate other TTMSS enol ethers in cascade aldol reactions with excellent levels of 1,2-Felkin and 1,3-syn-asymmetric induction. This is shown in the sequential addition of acetaldehyde- and acetophenone-derived TTMSS enol ethers to 2-phenylpropanal (Scheme 1.26) [51, 52]. On the basis of the open-chain model proposed by Evans ((1) in Scheme 1.24), the rationale for the observed 1,3-syn induction of β-OTTMSS aldehydes considers that the size of this silicon-protecting group now dictates that the carbonyl group is antiperiplanar to the OTTMSS substituent and far from the R1 group, which avoids unfavorable steric interactions. Therefore, conformation XVIII is responsible to the predominant formation of the 3,5-syn aldol diastereomer.
Scheme 1.26 Mukaiyama aldol reactions of tris(trimethylsilyl)silyl enolsilanes.
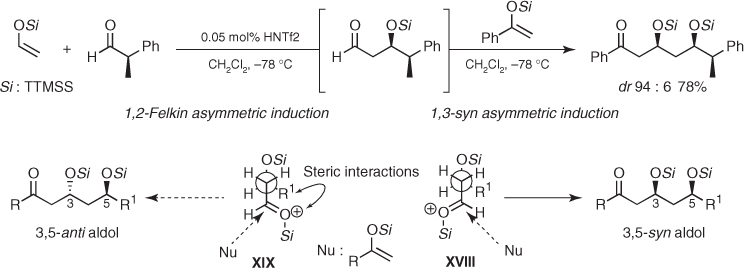
As discussed in Section 1.3.6, this chemistry combined with lithium-mediated aldol reactions from β-OTTMSS methyl ketones provides new entries to efficient synthesis of natural products.
The high asymmetric induction imparted by α- and β-substituted aldehydes documented in previous sections raises the prospect that there might exist intrinsic stereochemical relationships between both substituents that are either mutually reinforcing or opposing. This is particularly true for α-methyl β-alkoxy aldehydes.
Indeed, the stereochemical outcome of Mukaiyama aldol additions to these chiral aldehydes under nonchelation conditions can be easily interpreted by invoking the 1,2-Felkin and 1,3-anti asymmetric induction provided by the α-methyl and the β-oxygenated substituent, respectively. Hence, it is not surprising that the BF3-mediated additions of silyl enol ethers (42) to α-methyl-β-OPMB aldehydes (63) and (64) largely depend on the relative configuration of the aldehyde (Scheme 1.27). As the influences of the α-methyl and the β-OPMB substituents match in anti-substituted aldehyde (63), Felkin aldols (65) were virtually obtained as a single diastereomer, whereas syn-substituted aldehyde (64), in which those factors are opposing, gave mixtures of aldols (67) and (68) in variable diastereomeric ratios [53].
Scheme 1.27 Mukaiyama aldol additions to α-methyl β-alkoxy aldehydes.
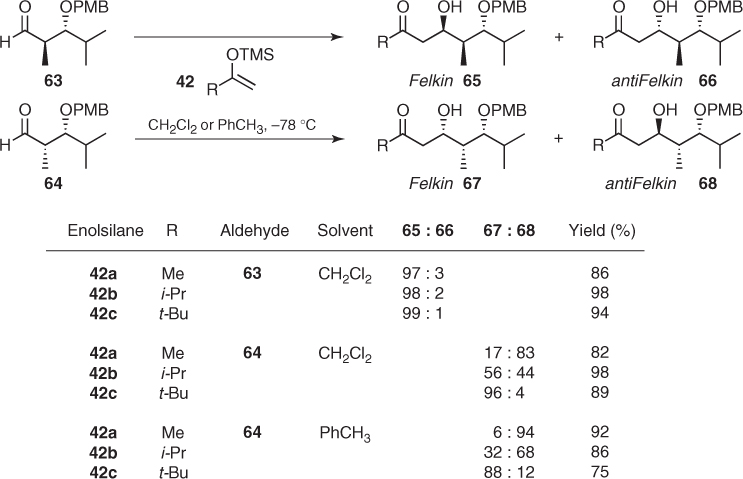
Interestingly, additions to 64 showed a turnover in carbonyl face selectivity (from Felkin to anti-Felkin) on decreasing the size of the enolsilane group R. This implies that the β-stereocenter becomes the dominant control element in the reactions with sterically nondemanding enolsilanes. Moreover, a decrease in the solvent polarity produced an increase of anti-Felkin diastereomer (68), which means that 1,3-induction is enhanced relative to 1,2-induction in nonpolar media. After a seminal analysis of these stereochemical data, Evans concluded that 1,2-stereoinduction is found in those reactions proceeding through an antiperiplanar open transition state such as XX, while dominant 1,3-stereoinduction is manifest from a synclinal open transition state such as XXII. Then, a subtle balance of stereoelectronic effects and steric interactions between the Lewis acid and the R group of the nucleophile on transition states XX–XXIII represented in Scheme 1.28 determines the stereochemical outcome of these reactions [53].
Scheme 1.28 Evans models for Mukaiyama aldol additions to syn α-methyl β-alkoxy aldehydes.
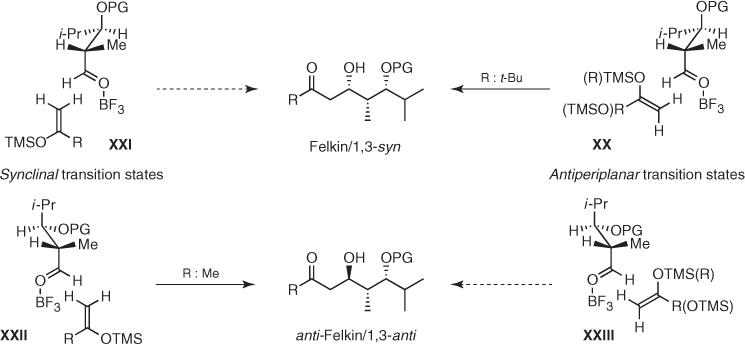
The consistently high stereocontrol provided by Mukaiyama aldol reactions of anti α-methyl β-oxygenated aldehydes has been successfully used in the synthesis of many natural products [54]. Evans reported that BF3-mediated vinylogous aldol addition of Chan's diene (52a) to chiral aldehyde (69) in toluene at − 90 °C gave β-hydroxy ketone (70), an advanced intermediate in the total synthesis of callipeltoside A (71), with excellent stereocontrol (dr > 95 : 5) in 88% yield ((1) in Scheme 1.29) [55]. In turn, Paterson developed a highly efficient coupling of chiral silyl enol ether (72) and elaborate α-methyl β-oxygenated aldehyde (73) to obtain aldol (74) as a single diastereomer in 91% yield, which was easily converted into preswinholide A (75) ((2) in Scheme 1.29) [56], the monomeric seco acid of swinholide A [57, 58]. Finally, the conversion of methyl ketone (76) into the corresponding silyl enol ether and subsequent BF3-mediated addition to aldehyde (77) in the presence of 4 Å molecular sieves allowed Roush to generate aldol (78), the penultimate precursor of bafilomycin A1 (79), with a high diastereoselectivity (dr > 95 : 5) in 72% yield ((3) in Scheme 1.29) [59].
Scheme 1.29 Use of asymmetric induction imparted by anti α-methyl β-oxygenated aldehydes in Mukaiyama aldol reactions in the synthesis of natural products.
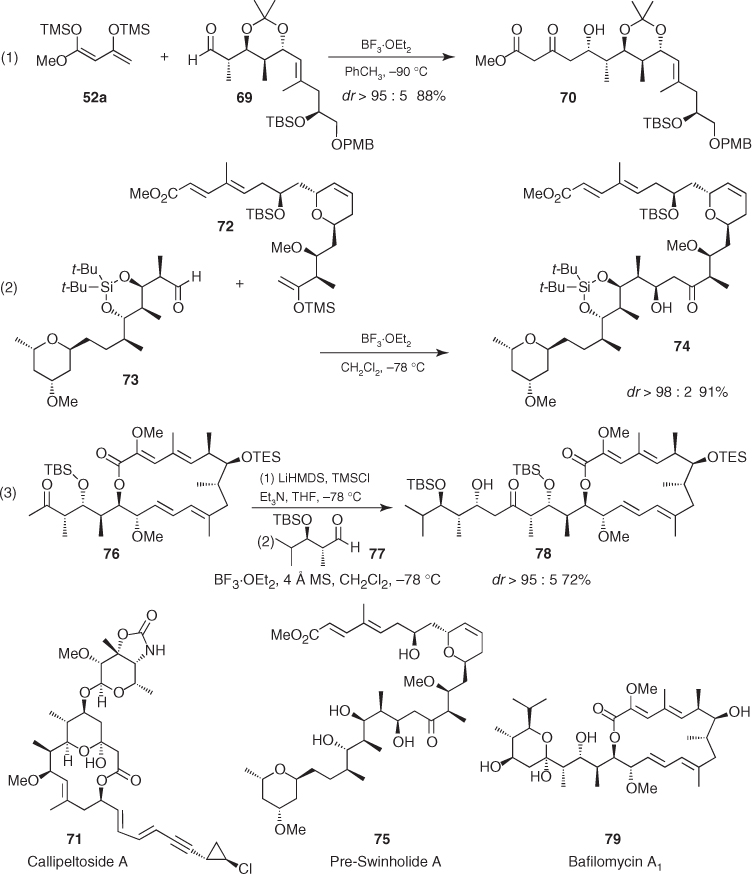
Parallel Mukaiyama aldol additions to syn-α-methyl β-oxygenated aldehydes have also been applied to the synthesis of natural products, but the uncertainty of their stereochemical outcome and the poorer diastereoselectivities often provided by these aldehydes have restricted their use compared to their anti counterparts. Remarkably, most of the examples found in the literature about these transformations involve Felkin-like reactions leading to all syn aldols. For instance, Floreancig reported the preparation of Felkin aldol (82) in excellent yield as an inseparable 86 : 14 mixture of diastereomers by addition of sterically hindered silyl enol ether (80) to chiral syn-α-methyl β-silyloxy aldehyde (81) ((1) in Scheme 1.30) [60, 61]. Furthermore, Kalesse described one of the few examples of substrate-controlled Mukaiyama aldol reaction in which the 1,3-induction of the β-oxygenated stereocenter prevailed over the Felkin induction imparted by the α-stereocenter [62]. This engaged the silyl enol ether (42a) addition to syn-aldehyde (83), which afforded aldol (84) with excellent diastereoselectivity (dr 97 : 3) in 86% yield ((2) in Scheme 1.30) [63].
Scheme 1.30 Asymmetric induction imparted by syn α-methyl β-oxygenated aldehydes in Mukaiyama aldol reactions.
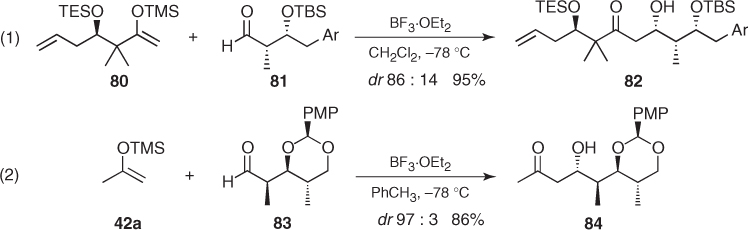
Following these analyses, π-facial selectivity of chiral aldehydes possessing α- and β-heteroatoms could be expected to arise from the summation of the inductions imparted by both substituents. Unfortunately, α,β-bisalkoxy aldehydes do not fulfill such expectations. Mukaiyama aldol additions to syn-α,β-bisalkoxy aldehydes under nonchelating conditions are too reliant on the hydroxyl protecting groups and the steric encumbrance of the enolsilane and usually proceed with poor stereocontrol. The same occurs to anti diastereomers, but anti-configured α-OBn β-OTBS aldehyde (85) exhibited uniformly high selectivities toward 3,4-anti aldols (86) irrespective of the steric hindrance of silyl enol ethers (42) ((1) in Scheme 1.31) [38]. A similar trend was observed for α-amino β-alkoxy aldehydes [64]. In this case, addition of silyl enol ether (42c) to anti α-N-BnTs β-OTBS aldehyde (87) afforded 3,4-anti aldol (88) as a single diastereomer in 92% yield ((2) in Scheme 1.31).
Scheme 1.31 Asymmetric induction imparted by chiral aldehydes possessing α- and β-heteroatoms in Mukaiyama aldol reactions.
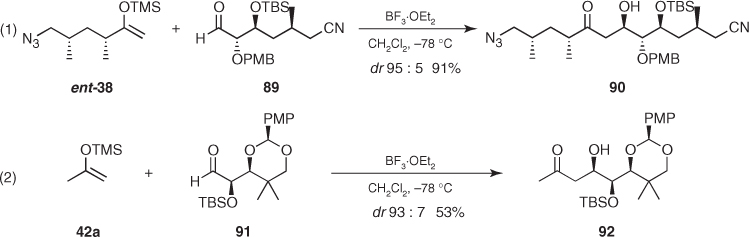
The highly diastereoselective aldol reaction of silyl enol ether (ent-38) and anti-aldehyde (89) affording anti aldol (90) is consistent with these features ((1) in Scheme 1.32). Moreover, alternative patterns occasionally proceed with excellent levels of diastereoselectivity. For instance, Paterson reported that silyl enol ether (42a) and syn-aldehyde (91) participated in a highly diastereoselective reaction (dr 93 : 7) toward all syn-aldol (92), thus indicating that the steric effect of the large alkyl group overrode any electronic stereocontrol from the oxygenated substituents ((2) in Scheme 1.32) [65, 66]. In spite of these successful examples, most of the stereocontrolled aldol reactions of α, β-bisalkoxy aldehydes rely on the use of lithium enolates (Section 1.3.5).
Scheme 1.32 Mukaiyama aldol reactions involving α,β-bisalkoxy aldehydes.
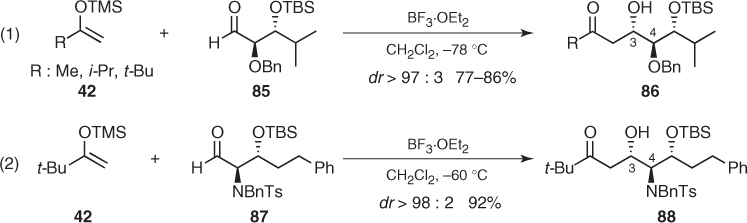
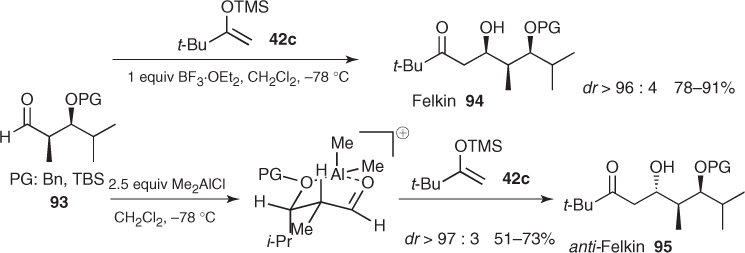
The stereochemical outcome of all these Mukaiyama aldol reactions involving α, β-disubstituted aldehydes can be dramatically affected if they are carried out under chelating conditions. Particularly, Evans established that the choice of the Lewis acid was crucial to the control of the additions to syn α-methyl β-OPG (PG: Bn, TBS) aldehydes (93) (Scheme 1.33) [67].3 Thus, BF3-mediated additions of silyl enol ether (42c) to these aldehydes provided Felkin aldols (94) in excellent diastereomeric ratios (dr > 96 : 4), whereas opposite anti-Felkin aldols (95) were obtained with the same level of diastereoselectivity by using Me2AlCl.4 Further theoretical and spectroscopic studies revealed that these transformations proceed through a cationic dimethylaluminum chelate that preferentially adopts a boat conformation and directs the attack of the nucleophile to the Si face of the C—O bond (Scheme 1.33). From a general point of view, these results demonstrate the exceptional chelating ability of this aluminum Lewis acid and expand the stereochemical control provided by these aldehydes [68, 69].
Scheme 1.33 Influence of Lewis acids on the stereochemical outcome of Mukaiyama aldol reactions of α-methyl β-alkoxy aldehydes.
Not surprisingly, the crucial role played by Lewis acids in Mukaiyama aldol reactions has stimulated the search for chiral ligands to dictate the stereochemical outcome of these transformations. The resultant chiral Lewis acids must increase the electrophilicity of aldehydes, create a suitable asymmetric environment around the carbonyl bond, and, as far as possible, facilitate catalytic turnover at the same time [5]. The following section describes how the intense efforts directed to this challenging objective have already provided highly enantioselective approaches, paying particular attention to those chiral Lewis acids used in the synthesis of natural products.
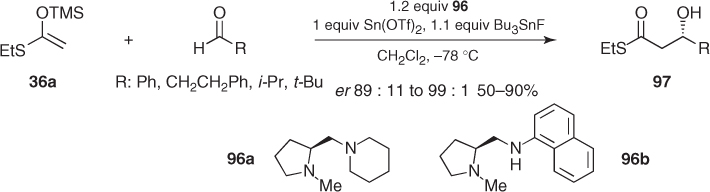
Mukaiyama and Kobayashi reported the first chiral complexes to provide stereocontrolled acetate Mukaiyama aldol reactions. These initially consisted of three pieces: Sn(OTf)2, an optically active diamine, and a tin(IV) additive. Thus, stoichiometric amounts of Sn(OTf)2, proline-derived diamines (96), and tributyltin fluoride promoted highly enantioselective additions of silyl ketene S,O-acetal (36a) to representative aldehydes, leading to the corresponding β-hydroxy thioesters (97) (Scheme 1.34) [70]. Application of these conditions to the TBS ketene acetal from benzyl acetate furnished similar results [71].
Scheme 1.34 Asymmetric Mukaiyama aldol reactions promoted by stoichiometric amounts of Sn(OTf)2 and proline-derived diamines.
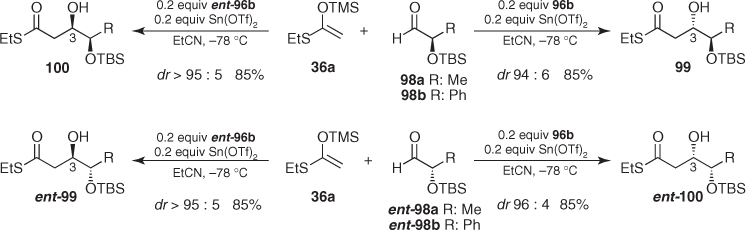
Remarkably, these additions were also successful when using catalytic amounts of Sn(OTf)2 and diamine (96b) without tributyltin fluoride provided that the reaction was carried out in propionitrile, and the enolsilane and the aldehyde were both added slowly to the reaction mixture [72]. The power of this methodology was demonstrated on the addition of 36a to chiral α-OTBS aldehydes (98) and (ent-98), in which the C3 stereocenter of aldols (99) and (100) was controlled by the diamine (96) regardless of the inherent diastereofacial preference of the chiral aldehydes (Scheme 1.35) [73, 74].
Scheme 1.35 Asymmetric Mukaiyama aldol reactions promoted by catalytic amounts of Sn(OTf)2 and proline-derived diamines.
Mechanistic analyses of this reaction suggested that such excellent stereocontrol might arise from the approach of the enolsilane to the less hindered face of the aldehyde in the highly ordered complex shown in Scheme 1.36 [75]. Regarding the catalytic turnover, the metal exchange reaction with TMSOTf to regenerate the catalyst was identified as a crucial step, because TMSOTf can promote an alternative achiral route (Scheme 1.36) that erodes the enantioselectivity if the silyl transfer is slow (kturnover ≈ kachiral). Thus, the need to minimize this side reaction led to the use of a more polar solvent such as propionitrile, which increases kturnover, and to the slow addition of both the enolsilane and the aldehyde to the reaction mixture.
Scheme 1.36 Mechanism for the Mulaiyama aldol reaction catalyzed by Sn(OTf)2 and a proline-derived diamine.
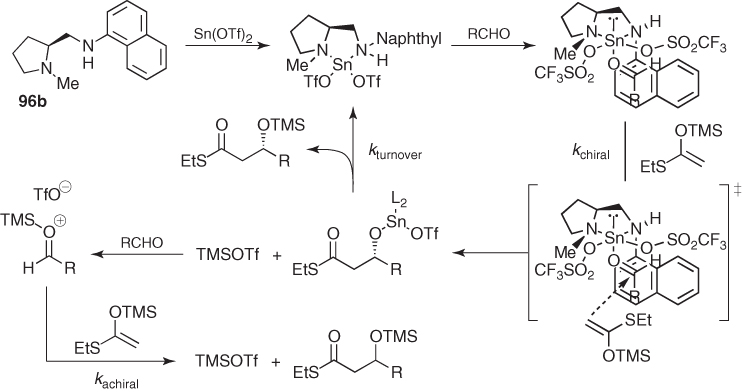
This methodology showed that catalytic asymmetric Mukaiyama aldol reactions were possible and could be used successfully in organic synthesis, but it suffered from the difficulty of handling Sn(OTf)2 [76] and the need for the reagents to be added to the reaction mixture slowly.
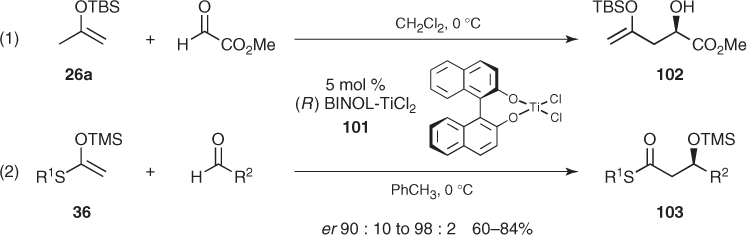
In the early 1990s, some chiral catalysts derived from the easily available BINOL ligand emerged as candidates to overcome such limitations. Mukaiyama was the first to use a BINOL-derived titanium–oxo complex to catalyze the addition of silyl ketene S,O-acetals to a limited number of aldehydes with modest stereocontrol [77]. This was followed by Mikami's findings on highly enantioselective ene and aldol reactions catalyzed by BINOL–titanium complex (101). Remarkably, addition of silyl enol ether (26a) to methyl glyoxylate afforded α-hydroxy ester (102) as a single enantiomer ((1) in Scheme 1.37) [78], whereas aldol reactions of silyl ketene S,O-acetals (36) and a wide array of aldehydes furnished thioesters (103) in good to high yields and enantiomeric ratios up to 98 : 2 ((2) in Scheme 1.37) [79].
Scheme 1.37 Asymmetric Mukaiyama aldol reactions catalyzed by BINOL-TiCl2.
Crossover experiments and analyses of the stereochemical outcome of related reactions led to the suggestion of a cyclic six-membered transition state for these transformations, wherein the oxygen of the activated carbonyl bond interacts with the transferring group, H or SiR3 for ene or aldol reactions, respectively (Scheme 1.38). Furthermore, the enantioselectivity was further improved by the addition of achiral ligands or by more bulky silicon groups, which suggests a more complex mechanism [80]. Irrespective of the mechanism, the synthetic potential of both transformations was demonstrated in two-directional ene processes [81] and aldol reactions involving chiral aldehydes by using a catalyst derived from BINOL and TiCl2(i-PrO)2 [82].
Scheme 1.38 Cyclic transition states for ene and aldol reactions.
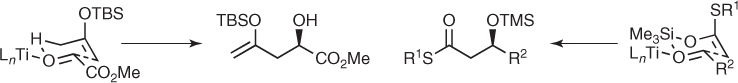
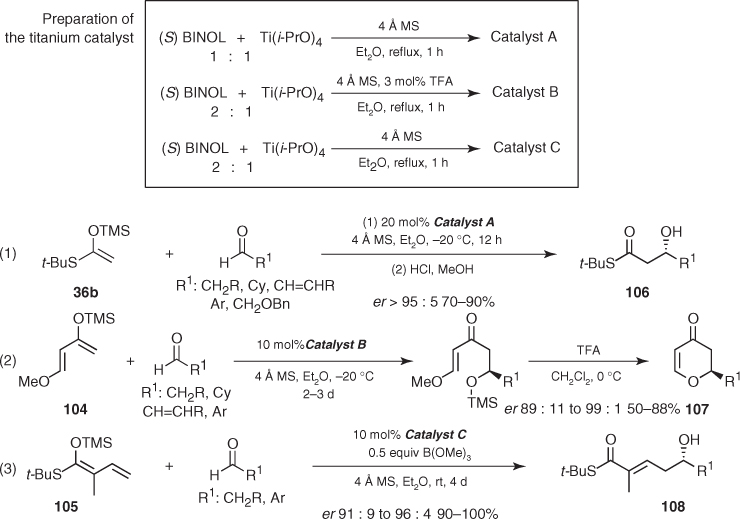
Keck reported a similar methodology in which the titanium catalyst was prepared by mixing BINOL ligand and Ti(i-PrO)4 [83]. The stereocontrol was again excellent, but Keck warned about the unknown structure of the catalytic species and emphasized the pronounced sensitivity of these reactions to BINOL–metal stoichiometry and the presence of other additives. Indeed, the catalytic species can be prepared according to several protocols adapted to different nucleophiles. Therefore, these have been applied to reactions from silyl ketene S,O-acetal (36b) [83], diene (104) [84, 85], and vinyl ketene S,O-acetal (105) [86] leading to β-hydroxy thioesters (106), dihydropyrones (107), and β-hydroxy α, β-unsaturated thioesters (108) in a highly enantioselective manner ((1–3) in Scheme 1.39).
Scheme 1.39 Asymmetric Mukaiyama aldol reactions catalyzed by BINOL/Ti(i-PrO)4.
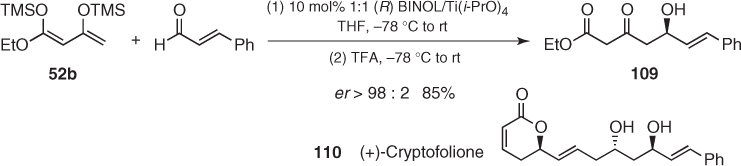
A thorough analysis and optimization of these transformations revealed the nonlinear effects and the dramatic influence of some additives [87, 88], which confirmed Keck's comments about the sensitivity of these reactions to structural and experimental modifications [89]. In spite of these limitations, these catalysts are an important tool for the stereocontrolled synthesis of natural products [90]. This is illustrated by the catalytic addition of Chan's diene (52b) to cinnamaldehyde affording aldol (109), an intermediate in the total synthesis of (+)-cryptofolione (110) described by Meshram (Scheme 1.40) [91].
Scheme 1.40 Synthesis of (+)-cryptofolione.
111ai4tert112113Scheme 1.41115112a114116Scheme 1.41O117118Scheme 1.41111bi4119Scheme 1.41