Natural Radioactivity
Dominic Larivière and Nicolas Guérin
Université Laval, Québec, QC, Canada
1 SUMMARY
The natural ionizing radiation present on our planet comes from many sources and generates most of the radioactivity that surrounds us. Primordial radionuclides, defined as radionuclides present since the formation of Earth about 4.6 × 109 years ago, play a critical role in our understanding of geological conditions on our planet over its whole history. Radioactivity initiated by the successive decay of 232Th, 235U, and 238U is essentially responsible for the presence of radioisotopes of lead, polonium, bismuth, astatine, radon, francium, radium, and protactinium on Earth. Because of their short half-lives with respect to Earth’s geologic timescale, the decay of these radionuclides is responsible for a significant portion of the radiation doses from natural radioactivity received by humans. Cosmic radiation, originating from both within and beyond our solar system, completes the list of significant modes of production of natural radioactivity in the atmosphere and the lithosphere. Minor sources of natural radioactivity (including radionuclides produced by cosmic neutron bombardments and spontaneous fission in uranium and thorium minerals) are known to generate quantities of transuranium isotopes and fission products, which are generally associated with the development of nuclear power.
Our actions as a society affect the distribution of natural radioactivity on Earth. Procedures that transform natural resources containing naturally occurring radioactive materials (NORM) generate by-products that have found their way into the environment. Technologically enhanced naturally occurring radioactive materials (TENORM) are produced by human activities such as fertilizer production and fossil fuel use, and are constantly modifying the distribution of radionuclides on Earth. Natural occurrences, including geologic and seismic events and forest fires, have also impacted the distribution of natural radioactivity.
2 NATURAL RADIOACTIVITY
2.1 Introduction
Radioactive substances are defined as materials that contain unstable atoms which produce ionizing radiation through nuclear rearrangement. Following the discovery of radioactivity in uranium sulfate by Becquerel in 1896, many scientists became involved in the isolation and characterization of radioactive substances. These investigations quickly led to the understanding that radioactive decays were not unique in nature, but produced various ionizing effects based on the type and energy of the radiation. The different nature of the ionizing radiation was linked to the type of rearrangement occurring within the radioactive nucleus, while the amount of energy generated by the decay was related to the amount of energy stored within the unstable nuclei.
On the basis of the observations of radioactivity made in the early 1900s, three types of nuclear rearrangements were recognized: α -, β −- and γ -decay. The structures of α - and β −-particles were identified as those of a charged helium atom 
 , respectively. It was later found that neutrinos (ν) and antineutrinos
, respectively. It was later found that neutrinos (ν) and antineutrinos  are also associated with β -decays. Sometimes after an α - or a β -decay, the newly formed nucleus is still in an excited and metastable state (represented bym). The decay from this excited state to a more stable state generates an electromagnetic wave with high energy (from 10 keV to several MeV), known as γ -radiation. Equations (1–3) illustrate the nuclear rearrangement leading to the production of α -, β -and γ -radiation, respectively.
are also associated with β -decays. Sometimes after an α - or a β -decay, the newly formed nucleus is still in an excited and metastable state (represented bym). The decay from this excited state to a more stable state generates an electromagnetic wave with high energy (from 10 keV to several MeV), known as γ -radiation. Equations (1–3) illustrate the nuclear rearrangement leading to the production of α -, β -and γ -radiation, respectively.
(1) 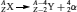
(2) 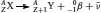
(3) 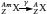
In (equations 1–3),  represents the isotope initiating the nuclear decay and is sometimes referred as the parent isotope. Decay or daughter products are represented in equations (1) and (2) as
represents the isotope initiating the nuclear decay and is sometimes referred as the parent isotope. Decay or daughter products are represented in equations (1) and (2) as  and
and  , respectively. Nowadays, many other types of nuclear rearrangements, such as electron capture, β +-decay and spontaneous fission, have been identified and have helped us better understand the fundamental nature of radioactivity.
, respectively. Nowadays, many other types of nuclear rearrangements, such as electron capture, β +-decay and spontaneous fission, have been identified and have helped us better understand the fundamental nature of radioactivity.
Nuclear rearrangements occur randomly within a group of radioactive atoms; however, statistically, it is possible to determine the number of disintegrations taking place within a defined period of time. Each radioisotope possesses a unique decay constant, represented as λ , which gives indications of the number of disintegrations per unit of time. The activity (A) of sample is therefore defined as
(4) 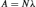
where N represents the number of atoms of a radioisotope present in the sample. Frequently, the decay constant is expressed as the half-life (t1/ 2), a concept defined as the time interval required for a certain number of radioactive atoms to decay by half. The relationship between λ and t1/2 is the following:
(5) 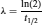
The remaining activity of a sample, for a time interval ranging from t 0 to t , can be calculated using the initial activity (A 0) of the sample using the following equation:
(6) 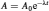
Radioactive isotopes (radioisotopes or radionuclides) are widely distributed on Earth, partitioned between the atmosphere and the lithosphere. Over 320 isotopes have been currently identified in nature. From that list, approximately 70 are known to have radioactive properties. While most elements found in nature have both stable and radioactive isotopes, elements with an atomic number higher than 83 only have the latter. The radioactivity on Earth consists of naturally produced radioisotopes and anthropogenic radioactive contamination initiated during the nuclear era (see Anthropogenic Radioactivity ; Civilian Nuclear Accidents).
This chapter covers the various modes of production of radioisotopes in the environment that are not the result of technological input. This type of discrimination between the radionuclides found in nature is however arbitrary, as some radionuclides found in the environment may have been naturally and anthropogenically produced. In addition, human actions and technologies can affect the environmental distribution of radioactivity without being part of its production. This point is covered later in this chapter. Natural radioactivity can be categorized into three distinct categories based on the origin of production of the radioisotope: cosmogenic radionuclides, primordial radionuclides, and radioactive decay series. This chapter describes the specificity and relevance of each category with respect to natural radioactivity.
2.2 Cosmogenic Radionuclides
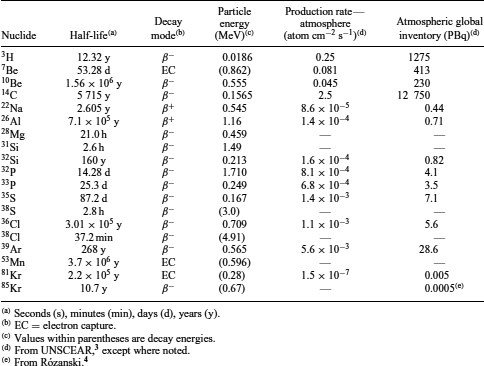
The Earth is constantly bombarded by cosmic radiation composed principally of high-energy particles emanating from extraterrestrial sources. Depending on its origin, the composition of the cosmic radiation varies greatly. Galactic cosmic radiation flux is typically composed of protons (87%), α -particles (11%), a few heavier nuclei with atomic numbers between 4 and 26 (∼1%), and some high-energy electrons (∼1%).1 In comparison, solar cosmic radiation, produced during solar energetic events, has a much higher proton composition (98%) and lower α -particle contribution (2%) and has no heavier nuclei or energetic electrons.2 Upon entering the Earth’s atmosphere, cosmic radiation interacts with the gaseous and particulate constituents to produce a variety of cosmogenic radioisotopes (Table 1).
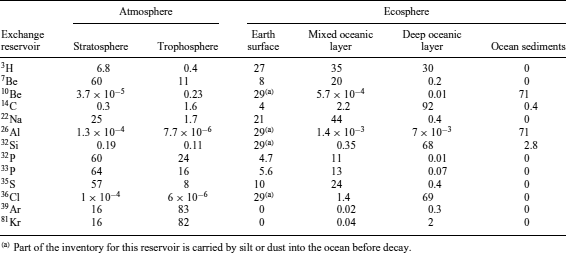
These interactions often generate a cascade of secondary particles such as protons and neutrons (Figure 1), which in turn will interact with target nuclei to produce additional cosmogenic nuclides. The largest number of nuclear transformations induced by cosmic radiation occurs within the Earth’s atmosphere, where most of the cosmic ray energy is dissipated. In contrast to what occurs within the atmosphere, the rate of nuclear transformations taking place at the Earth’s surface is several hundred times smaller;5 however, the omnipresence of some elements in soil and water relative to the atmosphere is responsible for the much higher partitioning of some cosmogenic nuclides (e.g., 36Cl) in the lithosphere (Table 2).
Most cosmogenic radionuclides are produced by one of the three nuclear rearrangement types involving cosmic particles: spallation, neutron capture, or muon capture. Spallation, a process where a nucleus splits into several lighter nuclei, proton, neutron, and muon after collision with a high-energy particle, is by far the most common mode of production of cosmogenic radionuclides in the atmosphere.6 Neutron and slow muon capture are far more common processes at the Earth’s surface, as the energy of the high-energy particles required for spallation is already dissipated in the upper layers of the atmosphere.
Table 1 Cosmogenic radionuclides
Table 2 Steady-state fractional inventory as a percentage of cosmogenic radionuclides in various exchange reservoirs. (Adapted from Lal and Peters5)
Table 3 and Figure 1 illustrate these production modes for three common cosmogenic radionuclides: 26Al, 14C, and 36Cl.
Table 3 Examples of typical nuclear processes leading to the production of cosmogenic radionuclides
The production rate (q i) of a nuclide i at a depth (h) from the upper boundary of the atmosphere, in either the atmosphere or the lithosphere, can be expressed as
(7) 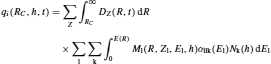
where RC represents the geomagnetic cutoff rigidity (the minimum energy a cosmic particle must have to create a cascade); Ml(R,Zl,El, h), the total differential multiplicity on the depth (h) in the atmosphere of active particle of type with an energy El and a charge Zle generated by a primary particle with charge Ze and rigidity R; Dz(R, t), the rigidity differential spectrum of primary cosmic radiation out of the atmosphere; σilk, the effective cross section of production of a cosmogenic radionuclide i by interaction between a particle of type l and a target nuclei of type k; and Nk, the concentration of this target nuclei. The kinetic energy of the primary particle (E(R)) can be defined as
(8) 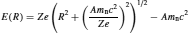
where A is the atomic number, m n is the rest mass of a nucleon and c is the speed of light. Using equations (7–8), it is possible to make estimations of the abundance of cosmogenic radiation in the atmosphere and lithosphere. While overall production of cosmogenic radionuclides has essentially been constant for over a thousand years, regional fluctuation must be expected as production rates are influenced by parameters such as energy, quantity, and the type of primary particles.7 Altitude and, to a smaller extent, latitude are also factors affecting the production of cosmogenic radionuclides (Figure 2). Cosmic particles proceeding through the atmosphere rapidly interact with atmospheric constituents, meaning that most radiation is produced at higher altitudes. Latitudinal effects are caused by the deflection of charged cosmic particles, by the magnetic field, away from the equator (0°) and toward the poles (90°).
Cosmogenic radionuclides present on Earth have a wide array of half-lives, ranging from less than a few minutes to several million years (Table 1). Shorter-lived radionuclides produced within the atmosphere usually decay before entering the ecosphere, but longer lived ones can reach the Earth’s surface. It has been estimated that cosmogenic radionuclides produced in the stratosphere have a residence time of approximately one year, except for those that are gaseous, which have longer residence time. After that time, they are generally transferred to the troposphere, where they reside for a much shorter period of time (typically 30–60 days).10 Finally, they are removed from the atmosphere by wet (rain) and dry (particulate) deposition to reach the Earth’s crust. The measurements of the exchange and mobility of cosmogenic radionuclides within the various environmental compartments of our planet are critical to our better understanding of many processes such as air–sea exchange and air circulation.11
Although radionuclide production through cosmic bombardment of atmospheric and terrestrial elements is the main mode of production of cosmogenic radionuclides on the Earth, cosmogenic radionuclides also come to Earth through extraterrestrial dust and meteorites that penetrate our atmosphere. Radioactivity in cosmic dust originates essentially from light radioisotopes, such as 7Be, 22 Na, 26Al, 46Sc, 48V, 51Cr, 53, 54Mn, 56, 57, 58, 60Co (see Cobalt), and 59Ni; the presence of much heavier cosmogenic radionuclides is generally attributed to meteorites.12 The upper limit of radioactivity attributable to dust and meteorites in the environment has been estimated at approximately 1.7 × 108 Bq.10 While this inventory might seem significant, its contribution is negligible in comparison to cosmogenic radioactivity originating from within the Earth’s atmosphere. As an example, 81Kr, one of the rarest cosmogenic radionuclides produced in the atmosphere, has a global inventory approaching 5 × 1012 Bq (see Xenon).3
The relative contribution of cosmogenic radionuclides to annual doses in the human population is illustrated in Figure 3. Cosmogenic radionuclides contribute to a mere 0.7% of the total dose received by humans through exposure to natural radioactivity. However, if doses linked to galactic and solar cosmic radiation are included, this percentage increases to almost 15%.13 While a number of cosmogenic radionuclides are known, the National Council on Radiation Protection and Measurements (NCRP) considers that only four of them (i.e. 14C, 3H, 22Na, and 7Be) contribute any measurable amount to the average dose received by humans.1
2.3 Primordial Radionuclides
Primordial radioactivity originates from radioisotopes which have half-lives comparable to the age of the Earth (4.5 × 109 years). Because of their long half-lives, primordial radionuclides have not decayed beyond the point of nondetection. Figure 4 illustrates the remaining activity as a function of time for six radionuclides with half-lives ranging from 107 to 1010 years. Radionuclides with shorter half-lives, exemplified by 236U (t1/2 = 2.37 × 107 years) in Figure 4, have activity levels that are negligible at the present time. Typically (see Uranium), a radionuclide present during the formation of the Earth but with a half-life of 108 years or shorter would currently have a remaining activity of less than 2.8 × 10−12% of its original activity. Therefore, all primordial radionuclides found on Earth have half-lives greater than 5 × 108 years.
Table 4 Primordial radionuclides outside of decay chains and with half-lives shorter than 1 × 1016 years
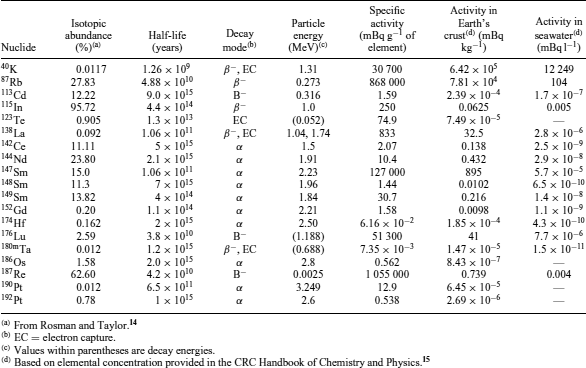
At least 17 naturally occurring single (nonseries) primordial radionuclides with half-lives between 109 and 1016 years have been identified (Table 4). Three radionuclides (232Th (see Thorium), 235U, and 238U) that initiate radioactive decay series, also of primordial nature, are discussed in Section 2.4. Some primordial radionuclides with extremely long half-lives (t1/2 > 1016 years) have been reported. They include 50V(1.4 × 1017), 76Ge (1.5 × 1021), 82Se (1.0 × 1020), 96Zr (3.9 × 1019), 100Mo (1.2 × 1019), 128Te (7.2 × 1024), 130Te (2.7 × 1021), 150Nd (1.7 × 1019), and 209Bi (1.9 × 1019). The half-lives provided here are the current values, but they might be reevaluated as precision in measurement techniques improve. Since primordial radionuclides have such long half-lives and relatively low elemental abundances, they are of little significance in terms of environmental concentration and dose, with the exception of 40K (see Potassium) and 87Rb (Figure 3 and Table 4).
One challenge in the detection of primordial radionuclides, other than 87Rb and 40K, resides in their low rate of disintegration. For example, in 1 g of samarium, one atom of 148Sm will decay every 700 s. As radiometric instruments rely on interactions between the ionizing radiations (e.g. α , β , and γ) generated from the nuclear decay and the detector, they are ill-suited for primordial radionuclide detection. For this reason, inorganic mass spectrometry is preferred to radiometric instrumentation for the detection of primordial radionuclides in the environment. Since mass spectrometry discriminates between isotopes on the basis of their atomic mass-to-charge ratio (m/z), not their rate of disintegration, the activity of the sample is not as critical for the determination of the radioisotopes as it would be for conventional radiometric methods. Based on the activity–mass relationship,16 which can be expressed mathematically as
(9) 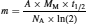
a radionuclide with a long half-life (t1/2) would have a much smaller activity (A) for an equivalent mass (m), based on a constant molar mass (M M), than a shorter lived one. Note that N A represents the Avogadro’s number. From equation 9, it is possible to determine the specific activity (S) of a radionuclide as
(10) 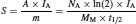
where I A is the isotopic abundance (Table 4). As noted by Kathren,10 it is unlikely that many more singly occurring primordial radionuclides will be discovered without significant advancements in measuring techniques for low-specific-activity radionuclides.
Independent of the challenge associated with their measurements, numerous dating applications based on primordial radionuclides found in the environment have emerged, especially with the development of new mass spectrometers, which are able to detect minute changes in isotopic ratios. Some radiochronometers and their applications in environmental dating are listed in Table 5.
Table 5 Primordial radiochronometers
2.4 Radionuclides from Natural Decay Series
The vast majority of the natural radioactivity detected on Earth and its related dose results from radionuclides belonging to the decay series which have primordial origins (Figure 3). While there were once four natural decay series present on the Earth, one of them has since completely decayed (Figure 5). These series are frequently characterized in terms of the mass number (A) of their constituents by the following expression:
(11) 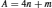
where n is the largest integer divisible into A and m is the remainder. For example, all the radioisotopes comprising the 4n series (Figure 6) have a mass divisible by four, with a remainder of zero. Series 4n + 2 and 4n + 3 are presented in Figures 7 and 8. All the nondecayed series (4n, 4n + 2 and 4n + 3) have a few common characteristics:
1. They originate from primordial radionuclides with significant half-lives (t1/2 > 108 year).
2. They possess a gaseous radioisotope of radon.
3. They end with a stable isotope of lead.
4. They decay via a series of α - and β -nuclear transformations.
The first two characteristics strongly dictate the presence and distribution of these series on Earth. The long half-life of the parents of these series is essential for their presence; otherwise they would now be extinct. A good example of the criticality of this characteristic is the 4n + 1 series, originating from 237Np (t1/2 = 2.14 × 106 years). This series has been created in the laboratory but cannot be found in the environment. However, the presence of 209Bi, the stable daughter of this series, in the environment indicates that it was once active on our planet. The presence of gaseous isotopes of radon, namely, thoron (220Rn), radon (222Rn), and acton (219Rn) for the 4n, 4n + 2 and 4n + 3 series, respectively, is largely responsible for the significant contribution of these series to the dose resulting from environmental sources.
The interrelated nature of the radionuclides that make up a decay series is perfect for the application of an activity steady-state concept, frequently referred to as equilibrium. At any point in time, the activity of a related radionuclide can be expressed as the difference between its production by its parent and its decay into a subsequent radionuclide. In a closed system, if two radionuclides linked through successive decay are allowed to decay for a sufficiently long period of time (several half-lives), three scenarios can occur. In the first scenario (Figure 9a), the half-life of the parent (i.e., 226Ra, t1/2 = 1599 years) is an order of magnitude longer than that of the daughter (222Rn, t1/2 = 3.823d), leading to a situation where the activity of both 226Ra and 222Rn will be equivalent.
This situation is called secular equilibrium , and is frequently used to estimate the activity of radionuclides that are challenging to detect, such as 210Pb (see Lead), which decays via a β -particle of low ionizing energy to 210Bi. If the scales of the half-life of the parent (214Bi, t1/2 = 19.7min) and the daughter (210Tl, t1/2 = 1.30 min) are similar, this scenario produces a situation where the two radionuclides will achieve a steady state in activity, but will both decay within the time frame of the experiment. This scenario is called transient equilibrium and is illustrated in Figure 9b. Finally, if the half-life of the parent (218Po, t1/2 = 3.04 min) is shorter than that of the daughter (214Pb, t1/2 = 26.9 min), equilibrium will never be reached (Figure 9c).
As stated previously, equilibrium can only be achieved in closed systems, where inflow and outflow of radionuclides are nil. This condition is rarely met in the environment, so deviations from equilibrium are frequently encountered in nature. Figure 10 illustrates this deviation from secular equilibrium (dashed line) for two pairs of the 4n + 2 series (210Pb/210Po and 226Ra/210Pb) in Canadian vegetation. Four mechanisms are recognized as responsible for the observed fractionation between radionuclides that make up the natural decay series: (1) solution and precipitation, (2) diffusion, (3) α -recoil, and (4) recoil-induced vulnerability to leaching.17 The precise determination of this fractionation has implications in numerous scientific fields, such as geology and oceanography, where it provides valuable information regarding the age of soil and water samples based on deviations from the expected equilibrium.18
2.4.1 The 4n Series (Thorium Series)
Initiated by the α -disintegration of 232Th (see Thorium), this decay series consists of six α -decays and four β -decays and concludes with 208Pb (Figure 6). This decay series is composed primarily of very short-lived radionuclides (t1/ 2 ≪ < 1 year), with the exception of 228Ra and 228Th, which have half-lives of 6.7 and 1.91 years, respectively. As a result of the significant half-life of 232Th, its activity has only decreased by approximately 20% since the formation of our planet (Figure 4). The United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR) has estimated that the 232Th decay series generates an annual effective dose of 0.34 mSv, of which 0.16 mSv is caused by external exposure and 0.18 mSv is an internal dose resulting from ingestion and inhalation.3 Almost 90% (0.16 mSv) of the dose is associated with the part of the series ranging from 220Rn to 208Tl. The remainder of the dose originates from the 232Th itself (0.003 mSv) and the part of the series ranging from 228Ra to 224Ra (0.013 mSv).
2.4.2 The 4n + 2 Series (Uranium Series)
Contrary to the 4n series, the 4n + 2 series (Figure 7), originating from 238U(see Uranium), has numerous radioisotopes with significant half-lives (≥15 years). This series has the unique characteristic that it has two uranium isotopes (234, 238U) which have been shown to exhibit disequilibrium in water and soil.20 From 238U, disintegrations occur in a series of eight α -decays and six β -decays to yield 206Pb. As 238U half-life is similar to the age of the Earth, roughly 50% of the 238U originally present has decayed (Figure 4).
The annual effective dose for the 4n + 2 series has been estimated as 1.34 mSv, with a contribution from external and internal exposure of 0.10 and 1.24 mSv, respectively.3 The impact of the uranium series on the internal dose can be divided into five disintegration regions, with the radionuclides from 222Rn to 214Po being the main contributors (1.1 mSv), and 210Pb, 210Bi and, 210Po being responsible collectively for approximately 0.12 mSv annually. The remainder of the dose originates from ingestion of 234, 238U, 230Th, and 226Ra (see Radium).
The large contribution to the internal dose from 222Rn progenies can be explained by the relatively long half-life of 222Rn (t1/ 2 = 3.825 days) compared to those of the other Rn isotopes, so this radioactive noble gas is able to permeate environmental mediums such as soil and water. It may then be inhaled before it decays, generating a significant internal dose.
The long half-life of 222Rn also provides enough time for the mixing of 222Rn in the atmosphere prior to its deposition into various environmental mediums such as water and soil, and facilitates the global distribution of its progenies in the environment (see Radon).
Determination of the sedimentation rates of lakes following unsupported 210Pb measurements (unsupported means that 210Pb is not generated by the disintegration of surrounding 226Ra) has been described and applied successfully to numerous watershed ecosystems.21
Disequilibrium is frequently amplified for radioisotopes from the 238U decay series, because of the relative mobility of 222Rn and its long half-life, which accentuates the fractionation between 226Ra and 210Pb into numerous environmental mediums. Differences in water solubility of complexes of 210Po and 210Pb enable, to a smaller extent, divergence from secular equilibrium. These two examples of variability in relative activities are illustrated in Figure 10.
2.4.3 The 4n + 3 Series (Actinium Series)
This series, originating from 235U, undergoes seven α -decays and four β -decays before ending with another stable lead (207Pb) isotope (Figure 8). Composed of three radioisotopes with half-lives longer than a year (235U, 231Pa, 227Ac), this series is responsible for the presence of quantities of actinium and protactinium (see Protactinium) of 5.5 × 10−3 and 1.4 ng kg−1, respectively, in the Earth’s crust.15 A significant fraction (∼98.7%) of 235U has already decayed since the formation of the planet because of its short half-life with respect to geological time (Figure 4). Because of the considerable decaying of 235U, its activity nowadays represents a mere 2% of the total activity from all naturally occurring uranium isotopes (234, 235, 238U), and it has very little impact on the effective dose.
2.5 Uncommon Natural Sources of Radioactivity Associated with Uranium
2.5.1 Neutron Capture from Uranium Isotopes
All the actinides found on Earth either have a half-life sufficient to survive total decay or are part of a nonextinct decay series. However, using very sensitive instrumentation, minute amounts of three actinides, 236U (t1/ 2 = 2.342 × 107 years), 237Np (t1/ 2 = 2.14 × 106 years), and 239Pu (t1/2 = 2.411 × 104 years), which do not adhere to these rules, have been discovered on Earth.22,23 These actinides, produced by neutron capture from 235U and 238U isotopes, are found in uranium-containing minerals. Table 6 shows the typical ratio values for these radionuclides in uranium ores.
2.5.2 Fission-Generated Radionuclides
Spontaneous fission in uranium ores occurs only rarely (238U: 5.45 × 10−5%) but leads to fission products. Tykva and Berg26 have reported 90Sr activity in the Earth’s crust equal to 50 PBq generated exclusively from spontaneous fission. This inventory is equivalent to a global concentration of 2μBq kg−1. 99Tc has also been reported in Canadian uranium ores at 99Tc/U ratio ranging from 1.5–800 × 10−12.24 The presence of fission products has also been reported in close proximity of the Oklo natural reactor in Gabon.27
2.6 Technologically Enhanced Naturally Occurring Radioactive Materials
While not a source of production of natural radionuclides per se, TENORM are responsible for the regional, national, and international distribution of many natural radionuclides in the environment. For this reason, they are briefly discussed in this section. TENORM, a term first coined by Gesell and Pritchard in 1975,28 describes the concentration of NORM modified to be used in consumer products and other human adaptations. Mining, fertilizer production, fossil fuel use, smelting, and water treatment and purification are examples of human activities known to modify the level of environmental radioactivity, especially in soil, water and air. The following paragraph highlights how some aspects of human activities magnify the level of natural radioactivity present.
2.6.1 Phosphate Fertilizer Production and Its Use in Agriculture
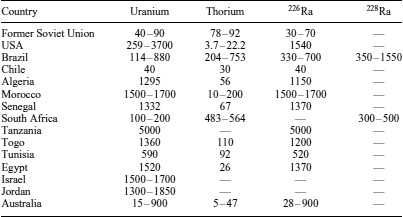
Agricultural phosphorus, essential for the development of crops, is obtained from phosphate rocks found in sedimentation formations (see Phosphorus). These formations contain trace quantities of uranium, thorium, and their progenies, incorporated in the structure of the mineral (Table 7). During the treatment of phosphate rocks with sulfuric acid to form phosphoric acid, radionuclides become partitioned between the products and the waste stream. Typically, 226Ra (∼80%), 232Th (∼30%), and 238U (∼14%) are left in the phosphogypsum produced through this approach.29 In the fertilizer, uranium and thorium concentrations are generally approximately 150% of their original concentration in the phosphate rock. Kathren30 has estimated that the distribution of fertilizers over American agricultural lands is equal to approximately 4 × 1013 Bq year−1 of 226Ra, 230Th, and 238U. These quantities will contribute to an annual equivalent effective dose of 10–20μSv, a small value in comparison with the contribution of natural radioactivity from decay series (Figure 3). Radionuclides dispersed on agricultural land via the spreading of fertilizers are redistributed in the environment by means of soil erosion and surface water runoff.
Table 6 Production reactions and isotopic ratios for uncommon naturally produced actinides
Table 7 Activity (Bq kg−1) of uranium, thorium, 226Ra, and 228Ra phosphate rocks from various locations 31
2.6.2 Fossil Fuel Use
Fossil fuels (e.g., oil, gas and coal), like most materials found in nature, contain traces of natural radioactivity which are released into the environment during the combustion process. In addition to various levels of uranium, thorium, and 40K, fossil fuel may still contain some quantities of 14C. One of the main radionuclides released from coal burning is 222Rn. Its production rate approaches 8 × 1012 Bq per 1000 MW generated.30 Quantities of 210Pb–210Po, and 40K (4 × 108 Bq) are also produced.30 Worldwide release of uranium and thorium into the atmosphere from coal burning has been estimated to be 3640 and 8960 tons, respectively.32 Of course, discharge of radionuclides in the atmosphere is strongly dependent on the combustion process and the efficiency of recovery of fly ash particles. Owing to the coal-burning process, concentrations of radionuclides in ashes are typically an order of magnitude higher than those in the original material (Figure 11). The National Council on Radiation Protection and Measurements (NCRP) has estimated that an American living within 80 km of a coal-fired electricity generating plant would receive an annual average effective equivalent dose rate of 0.3μSv year−1.
Specific activity of 14C in the environment has been significantly affected by human activities, especially those relying on fossil fuel. As quantities of 14C in coal and oil are dictated by the age of the material, the production of CO2 from those sources tends to have a much lower specific activity than that from recent carbon-based sources. This results in a reduction in the specific activity of atmospheric carbon in the environment, a phenomenon known as the Suess effect. The significance of the fossil fuel use in 14C activity can be observed for the period ranging from 1860 to 1950 (left-hand-side section in Figure 12). This effect has since been overshadowed by the large input of anthropogenic 14C from worldwide testing of atomic bombs (Figure 12, post-bomb period).
2.6.3 Building Materials
The presence of radioisotopes in building materials is the result of the natural occurrence of radioactivity in the raw material and the addition of industrial products, intermediates, or by-products such as coal ash, phosphogypsum, or furnace slags. The addition of industrial products to building materials is often motivated by the fact that it minimizes the use of resources and adds value to the materials. Table 8 presents the levels of radioactivity for 226Ra, 232Th, and 40K in some construction materials.
2.6.4 Water Treatment and Purification
Various processes are used to remove impurities and pollutants from water to make it potable. These processes include aeration, sand filtration, ion exchange, reverse osmosis, flocculation and sedimentation, coprecipitation, and softening through lime. Such actions accentuate the removal of heavy metals, dissolved salts, and radionuclides. Enhanced levels of radioactivity have been measured in flocculation sediment, sludges, spent ion-exchange resins, and reverse-osmosis cartridges used for water treatment (Table 9). As sediments and sludges are typically dried and disposed as landfill or land-spreading, remobilization of radioactivity in the environment is possible.
Table 8 Measured activity range (Bq kg−1) of 226Ra, 232Th, and 40K in building materials31
2.7 Geologic and Environmental Events Enhancing Levels of Natural Radioactivity
Many geological and environmental events can modify, as human activity does, the distribution of naturally occurring radionuclides. These events include volcanic action, droughts, floods, hydrothermal activity, ice melting, snow cap removal, earthquakes, and forest fire. In this section, we will briefly describe two of these processes and their impact on the redistribution of radionuclides in the environment.
2.7.1 Volcanic and Seismic Activities
Volcanic eruptions and earthquakes contribute to the remobilization of natural radioactivity by releasing volatile and gaseous radionuclides, previously secluded under the Earth’s crust, into the atmosphere. In volcanic emissions, radioactive isotopes of K, Pb, Bi, Po, and Rn have been measured.35 If radon is excluded, relative concentrations of the radionuclides mentioned above tend to correlate with the boiling points of their metal counterparts. Polonium is the most predominant in volcanic plumes, followed by bismuth, lead, and potassium. Ratios of 210Po/210Pb and 210Bi/210Pb of 56 and 3, respectively, have been measured in gaseous emissions from African volcanoes,36 clearly highlighting the high fractionation (ratio different from 1) induced by the extreme heat. Allard et al. 36 have estimated that the daily release of 210Po by the Erta ‘Ale volcano in Ethiopia reaches 3 × 109 Bq. For this reason, some research teams 37,38 have proposed that plumes of air contaminated by 210Po and 222Rn could be used as sensitive indicators of young volcanic events.
Table 9 Levels of radioactivity for a specific radionuclide (in parentheses) in selected water treatment wastes (from 3.8 × 105 L of water treated)31
| Treatment method |
Level of radioactivity (Bq l−1) |
| Coagulation/filtration (U) |
148 |
| Lime softening (Ra) |
20.6 |
| Ion exchange (Ra) |
7.7 |
| Reverse osmosis (U) |
11.8 |
Another source of release of radioactivity in the environment is linked to seismic activities. Hauksson and Goddard39 have noted significant fluctuations in 222Rn content in air collected over Iceland during seismic activities ranging in magnitude from 1.0 to 4.3. Igarashi et al. 40 observed a rise in 222Rn content in groundwater, approaching a 10-fold increase compared to the typical concentration (∼20 Bq l−1), days before an earthquake of magnitude 7.2 rocked Kobe, Japan, on January 17, 1995. While the origin and mechanisms of observed radon anomalies and their relationship to earthquakes are poorly understood, scientists have proposed that 222Rn monitoring could be used as an early warning signal.
2.7.2 Forest Fires
The release of quantities of naturally occurring radionuclides can also be attributed to biomass burning. Among the radionuclides redistributed from fires, radon progenies (210Pb, 210Bi and 210Po) represent the largest source.41 As for volcanic eruptions, their release in the atmosphere is favored because of their relatively low boiling temperatures. While typical atmospheric concentrations of 210Po and 210Pb are estimated to be around 37μBq m−3 and 370 μBq m−3,1 concentrations as high as 100 times these values have been detected in air samples collected during fire events.42 The nature of the fire, which will impact its temperature, will greatly affect the quantities of radionuclides released into the atmosphere. Lecloarec et al. 42 have reported that smoldering fires, where temperatures do not exceed 300°C, tend to release smaller amounts of radioactivity than flaming fires, where the temperature greatly exceeds 600°C. Quantities of 14C and 40K, incorporated as part of carbon and potassium intake by plants, can also be released into the atmosphere, contributing to the redistribution of radioactivity in the environment.
2.8 Conclusion
Natural radioactivity is an intrinsic part of life on Earth. Whether it originates from cosmic radiation, has been present on Earth since its formation, or results from a decay series, it is part of every medium of our environment. Natural radionuclides have a wide range of half-lives and environmental distribution, making them perfect tracers for dating and understanding the complex physico-chemical processes constantly reshaping our environment. With the exception of radiation exposure for medical purposes, natural radioactivity is responsible for the majority of the dose received by humans.
3 GLOSSARY
Anthropogenic radioactivity: Radioactivity generated by human nuclear activities.
β +-decay: Via this type of decay a proton in the nucleus turns into a neutron, a positron and a neutrino.
Effective cross-section: The effective cross-sectional area that an atom of an isotope presents to neutron scattering and absorption.
Effective dose: Sum of equivalent doses, weighted by the appropriate tissue weighting factors, in all the tissues and organs of the body.
Electron capture: Nuclear process during which unstable atoms gain stability. During electron capture, an electron in an atom’s inner shell is drawn into the nucleus where it combines with a proton, forming a neutron and a neutrino.
Fission-generated Radionuclides: Any radioactive nuclide resulting from the fission of a nucleus.
Half-life: Time required for a substance to lose half of its radiologic activity.
Muon capture: This type of nuclear reaction involves the capture of an unstable elementary particle (called muon) by a proton, usually producing a neutron and a neutrino, and occasionally a gamma photon.
Nucleon: Common name for a constituent particle of the nucleus.
Progeny: The decay product resulting after a radioactive decay or a series of radioactive decays.
Radiochronometer: Pair of isotope, including at least one that is radioactive, from which an age can be extrapolated from their current activity.
Radioisotope: Isotope that is unstable and release ionizing radiation.
Recoil: The motion gained by a particle as a result of its emission of another particle.
Residence time: The time during which radioactive material remains in a compartment of the environment following its natural or anthropogenic introduction in this compartment.
Secular equilibrium: Radioactive equilibrium in which the parent has such a long half-life compared to its progenies that there has been no appreciable change in the activity of the parent by the time the progenies have reached radioactive equilibrium.
Spallation: A nuclear reaction typically involving the collision of high-energy cosmic rays with an atom.
Spontaneous fission: Nuclear fission which occurs without the addition of particles or energy to the nucleus
Suess effect: The term refers to the dilution of the 14C/C ratio in atmospheric CO2 by the admixture of fossil-fuel produced CO2 depleted of its carbon-14.
Thermal neutrons: Neutrons in thermal equilibrium with the ambient medium.
Transient equilibrium: When the half-life of the parent radionuclide is slightly longer or about the same as the half life of the progeny, eventually equilibrium is reached. At this equilibrium, the total activity then decays at about the same rate as the original radionuclide.
4 ACKNOWLEDGMENTS
The authors would like to thank C. Bedwin and Dr. M.-E. Rousseau for their valuable comments during the completion of this chapter.
5 RELATED ARTICLES
Anthropogenic Radioactivity; Civilian Nuclear Accidents; Lead; Protactinium; Radon; Thorium; Uranium.
6 ABBREVIATIONS AND ACRONYMS
NCRP = National Council on Radiation Protection and Measurements; NORM = naturally occurring radioactive materials; TENORM = technologically enhanced naturally occurring radioactive materials.
7 REFERENCES
1. National Council on Radiation Protection and Measurements, ‘Exposure of the Population in the United States and Canada from Natural Background Radiation’ ; NCRP-94; Bethesda, MD (USA), 1987, 217.
2. J. N. Goswami, R. E. McGuire, R. C. Reedy, D. Lal, and R. Jha, J. Geophys. Res.-Space Phys. , 1988, 93 (A7), 7195.
3. United Nations Scientific Committee on the Effect of Atomic Radiation, ‘Sources, Effects and Risks of Ionizing Radiation’, United Nations Scientific Committee on the Effect of Atomic Radiation, New York, 1988.
4. K. Rózanski, Environ. Int. , 1979, 2 (3), 139.
5. D. Lal and B. Peters, Cosmic Ray Produced Radioactivity on the Earth, in ‘Encyclopaedia of Physics’, ed. Flugge, S., Springer-Verlag, Berlin, 1967, p. 551.
6. D. Lal, Annu. Rev. Earth Planet. Sci. , 1988, 16 , 355.
7. L. I. Dorman, ‘Cosmic Rays in the Earth’s Atmosphere and Underground’, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2004, p. 862.
8. J. Masarik and J. Beer, J. Geophys. Res. , 1999, 104 (D10), 12099.
9. J. C. Gosse and F. M. Phillips, Quat. Sci. Rev. , 2001, 20 (14), 1475.
10. R. L. Kathren, ‘Radioactivity in the Environment: Sources, Distribution, and Surveillance’, Harwood Academic Publishers, New York, 1984, p. 397.
11. D. Lal, Curr. Sci. , 1991, 61 (11), 744.
12. A. W. Klement Jr, Natural Sources of Environmental Radiation, in ‘CRC Handbook of Environmental Radiation’, ed. A. W. Klement Jr, CRC Press, Boca Raton, 1982, p. 5.
13. R. Michel, Radiochim. Acta , 1999, 87 (1–2), 47.
14. K.J. R. Rosman and P. D. P. Taylor, Pure Appl. Chem. , 1998, 70 (1), 217.
15. D. R. Lide, ed., ‘CRC Handbook of Chemistry and Physics’, CRC Press, Boca Raton, 2005.
16. D. Lariviere, V. F. Taylor, R. D. Evans, and R. J. Cornett, Spectrochim. Acta Part B-Atomic Spectrosc. , 2006, 61 (8), 877.
17. M. Ivanovich, Radiochim. Acta , 1994, 64 (2), 81.
18. M. Ivanovich and R. S. Harmon, ‘Uranium Series Disequilibrium: Applications to Earth, Marine and Environmental Sciences’, 2nd edition, Clarendon Press, Oxford, 1992, p. 912.
19. S. C. Sheppard, M. I. Sheppard, M. Ilin, J. Tait, and B. Sanipelli, J. Environ. Radioact. , 2008, 99 (6), 933.
20. S. J. Goldstein, J. M. Rodriguez, and N. Lujan, Health Phys. , 1997, 72 (1), 10.
21. J. A. Robbins, Geochemical and Geophysical applications of radioactive lead, in ‘The Biogeochemistry of Lead in the Environment’, ed. J. O. Nriagu, Elsevier/North-Holland Biomedical Press, New York, 1978, p. 285.
22. R. C. Thompson, Radiat. Res. , 1982, 90 (1), 1.
23. K. M. Wilcken, L. K. Fifield, T. T. Barrows, S. G. Tims, and L. G. Gladkis, Nucl. Instrum. Methods Phys. Res. B , 2008, 266 (16), 3614.
24. D. Curtis, J. Fabryka-Martin, P. Dixon, and J. Cramer, Geochim. Cosmochim. Acta , 1999, 63 (2), 275.
25. W. A. Myers and M. Lindner, J. Inorg. Nucl. Chem. , 1971, 33 (10), 3233.
26. R. Tykva and D. Berg, ‘Man-Made and Natural Radioactivity in Environmental Pollution and Radiochronology’, Kluwer Academic Publishers, Dordrecht, the Netherlands, 2004, p. 416.
27. R. Bros, L. Turpin, F. Gauthier-Lafaye, P. Holliger, and P. Stille, Geochim. Cosmochim. Acta , 1993, 57 (6), 1351.
28. T. F. Gesell and H. M. Prichard, Health Phys. , 1975, 28 (4), 361.
29. M. B. Cooper, ‘Naturally Occurring Radioactive Materials (NORM) in Australian Industries —Review of Current Inventories and Future Generation’, Rep. ERS-006, EnviroRad Services, Beaumaris, Australia, 2003.
30. R. L. Kathren, Appl. Radiat. Isot. , 1998, 49 (3), 149.
31. International Atomic Energy Agency (IAEA), ‘Extent of Environmental Contamination by Naturally Occurring Radioactive Material (NORM) and Technological Options for Mitigation’, Technical Reports Series No. 419, Vienna, Austria, 2003.
32. A. Gabbard, Oak Ridge Nat. Lab. Rev. , 1993, 26 (3–4), 25.
33. C. Papastefanou, J. Radioanal. Nucl. Chem. , 2008, 275 (1), 29.
34. P. P. Tans, A. F. M. Dejong, and W. G. Mook, Nature , 1979, 280 (5725), 826.
35. M. Pennisi, M. F. Lecloarec, G. Lambert, and J. C. Leroulley, Earth Planet. Sci. Lett. , 1988, 88 (3 – 4), 284.
36. P. Allard, C. Oppenheimer, A. J. S. McGonigle, A. Aiuppa, J. S. Levine, M. J. Wooster, and V. Tsanev, Magma Supply Rate to Erta Ale Lava Lake (Afar) Inferred from Measured Volatile and Heat Fluxes, ‘European Geosciences Union 1st General Assembly, Nice, France’, 25 – 30 April, 2004; Nice, France, 2004, 06601.
37. C. C. Su and C. A. Huh, Geophys. Res. Lett. , 2002, 29 (5), 1.
38. E. Y. Nho, M. F. LeCloarec, B. Ardouin, and M. Ramonet, Tellus B-Chem. Phys. Meteorol. , 1997, 49 (4), 429.
39. E. Hauksson and J. G. Goddard, J. Geophys. Res. , 1981, 86 (NB8), 7037.
40. G. Igarashi, S. Saeki, N. Takahata, K. Sumikawa, S. Tasaka, Y. Sasaki, M. Takahashi, and Y. Sano, Science , 1995, 269 (5220), 60.
41. G. Lambert, M. F. Lecloarec, B. Ardouin, and B. Bonsang, Long-lived Radon Daughters Signature of Savanna Fires, in ‘Global Biomass Burning: Atmospheric, Climatic, and Biospheric Implications’, ed. J. S. Levine, MIT Press, Cambridge, 1991, p. 181.
42. M. F. Lecloarec, B. Ardouin, H. Cachier, C. Liousse, S. Neveu, and E. Y. Nho, J. Atmos. Chem. , 1995, 22 (1 – 2), 111.
Anthropogenic Radioactivity
Jerzy W. Mietelski
Institute of Nuclear Physics, Kraków, Poland
1 SUMMARY
The term anthropogenic radioactivity covers sources of radioactivity not present on the Earth before the nuclear era (the so-called artificial radioactivity) and technically enhanced natural radioactivity. However, this article is mainly devoted to problems related to artificial radionuclides. Many of the basic facts regarding artificial radionulcides were established in the 1930s and 1940s. The main facts about radioisotope classification, radioactive decay, and artificial radionuclide production are presented in this article. This includes a general overview on anthropogenic sources of radioactivity such as nuclear explosions, nuclear accidents, routine releases from nuclear reactors, releases due to nuclear weapon production programs or from nuclear fuel reprocessing installations, nuclear medicine, and the modern techniques used to measure radioactivity from these sources. A very general introduction to the concept of radioecology is also presented.
2 INTRODUCTION
2.1 Definition of Anthropogenic Radioactivity
Anthropogenic radioactivity consists of two main components. One is the radioactivity of naturally occurring radioactive materials (the so-called NORM) enhanced by humans (this feature is called TENR —technically enhanced natural radioactivity). The other, and perhaps the most important component of “anthropogenic radioactivity”, is called artificial radioactivity . TENR, by definition, is caused by human activities; among these, mining and industry are the most important. Uranium (see Uranium), thorium (see Thorium), polonium (see Polonium), radium (see Radium), potassium (40K (see Potassium143see TritiumTENRsee Natural Radioactivity