
Contents
Preface to the Revised Edition
Preface to the First Edition
1. Introduction
1.1 INTRODUCTION
1.2 INTERMOLECULAR INTERACTIONS
1.3 STRUCTURAL ORGANIZATION
1.4 DYNAMICS
1.5 PHASE TRANSITIONS
1.6 ORDER PARAMETERS
1.7 SCALING LAWS
1.8 POLYDISPERSITY
1.9 EXPERIMENTAL TECHNIQUES FOR INVESTIGATING SOFT MATTER
1.10 COMPUTER SIMULATION
FURTHER READING
2 Polymers
2.1 INTRODUCTION
2.2 SYNTHESIS
2.3 POLYMER CHAIN CONFORMATION
2.4 CHARACTERIZATION
2.5 POLYMER SOLUTIONS
2.6 AMORPHOUS POLYMERS
2.7 CRYSTALLINE POLYMERS
2.8 PLASTICS
2.9 RUBBER
2.10 FIBRES
2.11 POLYMER BLENDS AND BLOCK COPOLYMERS
2.12 DENDRIMERS AND HYPERBRANCHED POLYMERS
2.13 POLYELECTROLYTES
2.14 ELECTRONIC AND OPTO-ELECTRONIC POLYMERS
FURTHER READING
QUESTIONS
3 Colloids
3.1 INTRODUCTION
3.2 TYPES OF COLLOIDS
3.3 FORCES BETWEEN COLLOIDAL PARTICLES
3.4 CHARACTERIZATION OF COLLOIDS
3.5 CHARGE STABILIZATION
3.6 STERIC STABILIZATION
3.7 EFFECT OF POLYMERS ON COLLOID STABILITY
3.8 KINETIC PROPERTIES
3.9 SOLS
3.10 GELS
3.11 CLAYS
3.12 FOAMS
3.13 EMULSIONS
3.14 FOOD COLLOIDS
3.15 CONCENTRATED COLLOIDAL DISPERSIONS
FURTHER READING
QUESTIONS
4 Amphiphiles
4.1 INTRODUCTION
4.2 TYPES OF AMPHIPHILE
4.3 SURFACE ACTIVITY
4.4 SURFACTANT MONOLAYERS AND LANGMUIR-BLODGETT FILMS
4.5 ADSORPTION AT SOLID INTERFACES
4.6 MICELLIZATION AND THE CRITICAL MICELLE CONCENTRATION
4.7 DETERGENCY
4.8 SOLUBILIZATION IN MICELLES
4.9 INTERFACIAL CURVATURE AND ITS RELATIONSHIP TO MOLECULAR STRUCTURE
4.10 LIQUID CRYSTAL PHASES AT HIGH CONCENTRATIONS
4.11 MEMBRANES
4.12 TEMPLATED STRUCTURES
FURTHER READING
QUESTIONS
5 Liquid Crystals
5.1 INTRODUCTION
5.2 TYPES OF LIQUID CRYSTALS
5.3 CHARACTERISTICS OF LIQUID CRYSTAL PHASES
5.4 IDENTIFICATION OF LIQUID CRYSTAL PHASES
5.5 ORIENTATIONAL ORDER
5.6 ELASTIC PROPERTIES
5.7 PHASE TRANSITIONS IN LIQUID CRYSTALS
5.8 APPLICATIONS OF LIQUID CRYSTALS
FURTHER READING
QUESTIONS
6 Biological Soft Matter
6.1 INTRODUCTION
6.2 LIPID MEMBRANES
6.3 DNA
6.4 PROTEINS
6.5 POLYSACCHARIDES AND GLYCOPROTEINS
6.6 MACROMOLECULAR ASSEMBLIES
FURTHER READING
QUESTIONS
Numerical Solutions to QUESTIONS
Index

Copyright © 2007 John Wiley & Sons, Ltd, The Atrium, Southern Gate, Chichester,
West Sussex PO19 8SQ, England
Telephone (+44) 1243 779777
The right of Ian W. Hamley to be identified as the author of this work has been asserted in accordance with sections 77 and 78 of the Copyright, Designs and Patents Act 1988.
Email (for orders and customer service enquiries): cs-books@wiley.co.uk
Visit our Home Page on www.wileyeurope.com or www.wiley.com
All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London W1T 4LP, UK, without the permission in writing of the Publisher. Requests to the Publisher should be addressed to the Permissions Department, John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex PO19 8SQ, England, or emailed to permreq@wiley.co.uk, or faxed to (+44) 1243 770620.
Designations used by companies to distinguish their products are often claimed as trademarks. All brand names and product names used in this book are trade names, service marks, trademarks or registered trademarks of their respective owners. The Publisher is not associated with any product or vendor mentioned in this book.
This publication is designed to provide accurate and authoritative information in regard to the subject matter covered. It is sold on the understanding that the Publisher is not engaged in rendering professional services. If professional advice or other expert assistance is required, the services of a competent professional should be sought.
The Publisher and the Author make no representations or warranties with respect to the accuracy or completeness of the contents of this work and specifically disclaim all warranties, including without limitation any implied warranties of fitness for a particular purpose. The advice and strategies contained herein may not be suitable for every situation. In view of ongoing research, equipment modifications, changes in governmental regulations, and the constant flow of information relating to the use of experimental reagents, equipment, and devices, the reader is urged to review and evaluate the information provided in the package insert or instructions for each chemical, piece of equipment, reagent, or device for, among other things, any changes in the instructions or indication of usage and for added warnings and precautions. The fact that an organization or Website is referred to in this work as a citation and/or a potential source of further information does not mean that the author or the publisher endorses the information the organization or Website may provide or recommendations it may make. Further, readers should be aware that Internet Websites listed in this work may have changed or disappeared between when this work was written and when it is read. No warranty may be created or extended by any promotional statements for this work. Neither the Publisher nor the Author shall be liable for any damages arising herefrom.
Other Wiley Editorial Offices
John Wiley & Sons Inc., 111 River Street, Hoboken, NJ 07030, USA
Jossey-Bass, 989 Market Street, San Francisco, CA 94103-1741, USA
Wiley-VCH Verlag GmbH, Boschstr. 12, D-69469 Weinheim, Germany
John Wiley & Sons Australia Ltd, 42 McDougall Street, Milton, Queensland 4064, Australia
John Wiley & Sons (Asia) Pte Ltd, 2 Clementi Loop #02-01, Jin Xing Distripark, Singapore 129809
John Wiley & Sons Ltd, 6045 Freemont Bovd, Mississauga, Ontario L5R 4J3, Canada
Wiley also publishes its books in a variety of electronic formats. Some content that appears in print may not be available in electronic books.
Anniversary Logo Design: Richard J. Pacifico
Library of Congress Cataloguing-in-Publication Data
Hamley, Ian W.
Introduction to soft matter: synthetic and biological self-assembling materials / Ian W. Hamley. – Rev. ed.
p. cm.
Includes bibliographical references and index.
ISBN 978-0-470-51609-6 (cloth)
1. Polymers–Textbooks. 2. Colloids–Textbooks. 3. Micelles–Textbooks. 4. Liquid crystals–Textbooks. I. Title.
QD381.H37 2007
547′.7-dc22 2007025456
British Library Cataloguing in Publication Data
A catalogue record for this book is available from the British Library
ISBN 978-0-470-51609-6 (H/B)
978-0-470-51610-2 (P/B)
Preface to the Revised Edition
The field of soft matter science remains an exciting and fast developing one. Since the first edition of this book there have been advances in several areas and I felt it appropriate to update the book to reflect this. Several other books in the field have also been published recently (as cited in the revised bibliography sections herein), although most, fortunately, do not overlap too much with mine! The main addition to this text is a new chapter on Biological Soft Matter, which adds significantly to the rather cursory discussion of proteins and DNA in the previous edition and introduces some quite cutting-edge topics. I have also added new sections to Chapter 2 on advanced polymeric materials to discuss dendrimers, polyelectrolytes, and electronic and optoelectronic polymers, albeit briefly. The section about the thermodynamics of micellization in Chapter 4 has also been revised. I thank my students who have sat my Physical Chemistry of Soft Matter courses over the years in various guises for critical comments on the previous treatment, and John Wiley & Sons, Ltd for supporting me in this endeavour.
Ian W. Hamley
School of Chemistry,
University of Reading, UK
Preface to the First Edition
This book is largely intended to provide an introduction to colloid chemistry, but I have used the term ‘soft matter’ to indicate a unified subject that includes aspects of liquid crystal and polymer science not found in existing textbooks on colloid chemistry. General textbooks of physical chemistry either do not cover colloid chemistry at all or fail to give it the space it deserves. There are fewer than a handful of recent books that give a broad coverage of the physical chemistry of soft materials, and these are written at an advanced level. For both these reasons, and also given that existing introductory colloid chemistry texts are mostly getting rather long in the tooth, I felt that a new book in the area offering up-to-date and unified coverage, would be valuable both to students and researchers.
The book has been written primarily for undergraduates taking physical chemistry courses. In Leeds it is a companion book to the final year module ‘Physical Chemistry of Condensed Matter’. I hope it will also be of interest to students of physics and materials science taking courses on colloids, polymers, soft condensed matter or complex fluids. It should also serve as a useful introduction and reference for researchers in these areas.
I wish to thank my editors at Wiley for assistance in the production of the book.
IWH
School of Chemistry,
University of Reading, UK
Mankind has exploited matter in technology through the ages. For many millenia, we relied on materials like wood or metals that were subject to minimal processing to provide useful objects. It is only within a few minutes of midnight on the proverbial human evolutionary clock that materials have been engineered for ultimate applications based on a deep understanding of molecular properties. Considering substances that have been engineered in a controlled or tailored manner, the nineteenth century was the age of iron and steel. The twentieth century saw the development of new types of engineered materials, especially polymers, which in the form of plastics have, in many applications, usurped many of the traditional ‘hard’ materials. This is not to forget the emergence of an important class of inorganic material, semiconductors, in the second half of this century. These are, of course, the basis for the second industrial revolution, that of information technology. However, it seems fair to say that many properties of hard matter are now well understood whereas we are still on the learning curve with soft matter. For example, inspired by nature, we are only just beginning to be able to engineer complex structures formed by biopolymers or to exploit nanotechnology to make devices based on self-organization of polymers. In our new millennium it seems safe to predict the continued importance of soft materials, engineered in ways we can as yet only dream of.
The idea of a unified approach to ‘soft materials’ has only gained ground recently. It is an interdisciplinary subject, taking in aspects of physics, chemistry and materials science, but also of biochemistry or (chemical, mechanical) engineering in specific cases. A consequence of this interdisciplinarity is that, unfortunately, the subject is not considered in conventional textbooks on physics, physical chemistry or materials science, often being neglected entirely or covered in an inadequate manner. The purpose of this book is to ‘fill the gap’, by providing an up-to-date introductory summary of the thermodynamics and dynamics of soft materials. In each of the six chapters, the basic physical chemistry is covered first, prior to an outline of applications. The material is presented in a coherent fashion across the book. Equations have been kept to the minimum number that capture important relationships. Derivations are included, where they illustrate thermodynamical or statistical mechanical principles in action. The derivation of the Flory–Huggins theory in Section 2.5.6 or of the thermodynamics of micellar equilibria in Section 4.6.5 are good examples. Soft materials are important in many products, such as detergents, paints, plastics, personal care products, foods, clays, plastics and gels. Such uses of soft materials are exemplified throughout this book.
In this book we consider soft materials under the headings of polymers (Chapter 2), colloids (Chapter 3), amphiphiles (Chapter 4), liquid crystals (Chapter 5) and biological soft materials (Chapter 6). The distinctions between these systems are often not strong. For example, amphiphiles in solution and some aspects of polymer science are often considered in books on colloid chemistry. However, here we treat them separately since they are technologically important enough to merit detailed consideration on their own. The chapter on liquid crystals is in fact focused on one class of material, thermotropic liquid crystals, where phase transitions are thermally driven. However, a different class of liquid crystal phase is formed in amphiphile solutions, where concentration is also a relevant variable. These are termed lyotropic liquid crystal phases and are discussed in Chapter 4.
There are a number of texts that deal with aspects of the subjects covered in this book. General texts in the area include those by Evans and Wennerström, Hunter, Larson and Shaw (see Further Reading at the end of the chapter). Detailed textbooks for background reading on each topic are listed in the Further Reading section that follows each chapter. In Chapter 2, polymer science is outlined in a particularly concise form, and after the fundamentals are introduced, attention is paid to applications of polymers in the latter part of the chapter. There are quite a number of monographs concerned with colloids. However, many of these are not suitable for use as undergraduate textbooks. Thus, Chapter 3 fulfills a particularly useful function in providing an up-to-date introduction to the essential physical chemistry. Also emphasized are applications of colloids and colloids in everyday life, such as in foods. Chapter 4 summarizes the important aspects underpinning the self-assembly of amphiphiles, i.e. surfactants and lipids. The action of surfactants as detergents is also considered and the importance of lipids in cell membranes is discussed. Chapter 5 is concerned with thermotropic liquid crystals. Chapter 6 is focused on aspects of self-assembly of biological soft materials. Recommended texts for background reading on these subjects are listed in the Further Reading sections.
In this chapter, intermolecular forces that are the basis of self-assembly are considered in Section 1.2. Section 1.3 outlines common features of structural ordering in soft materials. Section 1.4 deals similarly with general considerations concerning the dynamics of macromolecules and colloids. Section 1.5 focuses on phase transitions along with theories that describe them, and the associated definition of a suitable order parameter is introduced in Section 1.6. Scaling laws are defined in Section 1.7. Polydispersity in particle size is an important characteristic of soft materials and is described in Section 1.8. Section 1.9 details the primary experimental tools for studying soft matter and Section 1.10 summarizes the essential features of appropriate computer simulation methods.
The term ‘soft’ matter originates from macroscopic mechanical properties. We mean here materials such as colloids, surfactants, liquid crystals, certain biomaterials and polymers in the melt or solution. Many soft materials can be induced to flow under certain conditions. This weak ordering results from the lack of three-dimensional atomic long-range order found in a crystalline solid. Nevertheless, there is always a degree of local order at least as great as that in a liquid. From the viewpoint of kinetic energy, a crude distinction between ‘soft’ materials and ‘hard’ ones can be made on the basis that the molecular kinetic energy for the former is close to kBT, whereas for the latter it is much less than kBT (when the temperature is near ambient). Here we consider the intermolecular forces responsible for the ordering of soft materials. Our purpose is not to provide a detailed description of these forces, since this is dealt with in many physical chemistry textbooks (for example Atkins, 2006). Here we briefly outline the essential results, especially in the context of self-assembly in soft matter, which is the subject of this book.
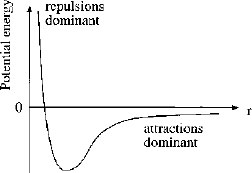
The forces between molecules are a balance of repulsive interactions at short distances and attractive interactions that predominate over larger length-scales. This is illustrated by the curve of potential energy as a function of intermolecular separation in Fig. 1.1. We will now consider the origin of the repulsive and attractive forces. Then we consider Coulombic forces since ions are present in solution in many colloid and surfactant systems, and in this case interactions between charged species predominate.
Figure 1.1 Typical curve of potential energy versus separation of two molecules or atoms. At short distances, repulsive interactions predominate, whilst attractive forces act over a longer range
Repulsive interactions are important when molecules are close to each other. They result from the overlap of electrons when atoms approach one another. As molecules move very close to each other the potential energy rises steeply, due partly to repulsive interactions between electrons, but also due to forces with a quantum mechanical origin in the Pauli exclusion principle. Repulsive interactions effectively correspond to steric or excluded volume interactions. Because a molecule cannot come into contact with other molecules, it effectively excludes volume to these other molecules. The simplest model for an excluded volume interaction is the hard sphere model. The hard sphere model has direct application to one class of soft materials, namely sterically stabilized colloidal dispersions. These are described in Section 3.6. It is also used as a reference system for modelling the behaviour of simple fluids. The hard sphere potential, V(r), has a particularly simple form:
(1.1) 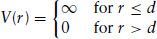
where d is the diameter of the hard sphere. The ordering of hard spheres depends only on their volume fraction. The phase diagram has been obtained by computer simulations and experiments on sterically stabilized colloid particles, as discussed in Section 3.6.
The hard sphere model is based on the excluded volume of spherical particles. An excluded volume theory has been developed to account for the orientational ordering of liquid crystal molecules, assuming them to be hard rods. This is the Onsager theory and its variants, outlined in Section 5.5.2. Excluded volume interactions influence the conformation of polymer chains. The conformation of an ideal chain is described by a random walk. However, in this case the chain can cross itself, i.e. it has no excluded volume. Under certain circumstances a polymer chain can behave as if this was the case (see Section 2.3.2). However, it is more usual for excluded volume interactions to lead to a self-avoiding walk, which produces a more extended conformation than that of a random walk (Section 2.3.2).
Because there are no attractive interactions in the potential, the hard sphere model does not describe the forces between molecules very well. More realistic potentials include an attractive contribution, which usually varies as − 1/r6 (as discussed shortly) as well as a repulsive term. The latter is chosen to vary as 1/rn, with n > 6, to ensure that repulsions dominate at short distances, n = 12 often being assumed. This combination of attractive and repulsive terms defines the Lennard–Jones (12,6) potential:
(1.2) 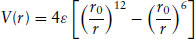
Here ε is the depth of the potential energy minimum and 21/6r0 is the intermolecular separation corresponding to this minimum. This potential has a form similar to that shown in Fig. 1.1. It is often used as a starting point for modelling intermolecular interactions, for example it can be chosen as the intermolecular potential in computer simulations (see Section 1.10). It is not completely realistic, though, because for example it is known that the 1/r12 form is not a good representation of the repulsive potential. An exponential form exp(−r/r0) is better because it reproduces the exponential decay of atomic orbitals at large distances, and hence the overlap which is responsible for repulsions.
Attractive interactions in uncharged molecules result from van der Waals forces, which arise from interactions between dipoles. A molecule has a dipole moment if it contains two opposite charges of magnitude q, separated by some distance r. Such a molecule is said to be polar. The dipole moment is then defined by μ = qr. Dipole moments of small molecules are usually about 1 debye (D), where 1D = 3.336 × 10−30 C m. Some molecules, such as H2O, possess a permanent dipole moment due to charge separation resulting from the electro-negativity of the oxygen atom. Dipolar molecules can also induce dipole moments in other molecules producing dipole–induced dipole forces. The potential energy between two dipoles can be calculated by summing up the Coulomb potential energy between each of the four charges. Recall that the Coulomb potential energy is given by
(1.3) 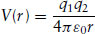
where r is the distance between charges q1 and q2 and ε0 is the vacuum permittivity.
Considering two parallel dipoles, the potential can be shown to vary as 1/r3. However, if the dipoles are freely rotating, the potential varies as 1/r6. Further details of the derivations of these functional forms are provided by Atkins (2006). Here we have only discussed the dependence of the potential on r; all prefactors are omitted. This type of relationship is an example of a scaling law, discussed in more detail in Section 1.7.
Most molecules are non-polar. It is evident, however, that there must be attractive van der Waals interactions between such molecules in order that condensed phases, such as those exhibited by liquid hydrogen or argon at low temperature, can exist. Molecules without a permanent dipole moment can possess an instantaneous dipole moment due to fluctuations in the atomic electron distribution. These fluctuating dipoles can induce dipoles which create a transient electric field that can polarize nearby molecules, leading to an induced dipole. Such induced dipole–induced dipole forces create dispersion interactions, also known as London interactions. It is again found that the potential varies as 1/r6. In other words, for both freely rotating dipole–dipole and induced dipole–induced dipole interactions, the attractive potential takes the form
(1.4) 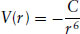
although the term C is different for the two types of interaction (further details are provided by Atkins, 1998, for example). This explains why the attractive contribution to the total potential is often taken to have the 1/r6 form, as in the Lennard–Jones (12,6) potential (Eq. 1.2) for example.
Coulombic forces dominate other interactions in systems containing ions. This is because the Coulombic potential energy falls off much more slowly (as 1/r, Eq. 1.1) than any dipole–dipole interaction. A typical Coulombic potential energy is ~250 kJ mol−1, whereas a van der Waals energy is about 1 kJ mol−1 or less.
An important distinction can be made between materials in which the structure comes from intermolecular ordering and those for which it is produced by the ordering of molecular aggregates. Many soft materials, such as colloids and micellar amphiphiles, belong to the latter class. A necessary condition for the formation of such aggregates is the existence of at least two components in the system. Often the second component is water and then hydrogen bonding interactions are important. In fact, hydrogen bonding is predominantly a type of dipole–dipole interaction, although there may also be some covalent character. For amphiphiles in solution, the hydrophobic effect drives the formation of micelles. The hydrophobic effect originates in the local structuring of water, which consists of a tetrahedral arrangement of hydrogen-bonded molecules. When an insoluble species such as a hydrocarbon is added to the water, this structure has to accommodate itself around each molecule, which produces a reduction in entropy. This is known as the hydrophobic effect. This structuring effect is reduced when the molecules assemble into micelles. The hydrophobic effect is discussed further in Section 4.6.5. We note here that since the hydrophobic effect has its origin in the entropy associated with local hydrogen bonding of water molecules, it ultimately depends on dipole–dipole forces.
Even in systems where structure results from molecular self-assembly into aggregates, it is forces between molecules that drive the self-assembly process, although these can be between molecules of different types. In one-component systems such as thermotropic liquid crystals, ordering can only result from forces between molecules of the same type. It is difficult to make a quantitative statement about the precise form of the potential for any soft material, other than observing that it will be some combination of repulsive short-ranged contributions and attractive long-range contributions.
There are both common and distinct features in the ordering of different types of soft material. The most important feature in common is that the ordering is generally intermediate between that of a crystalline solid and that of a liquid. This lack of crystalline order leads to the ‘soft’ mechanical response of the materials. There may, however, be partial translational and/or orientational order of molecules due to the formation of a mesophase by a thermotropic liquid crystal or an amphiphile in water. Polymer melts and solutions are also classified as ‘soft materials’, although there is no long-range translational or orientational order. However, these phases are distinguished from conventional liquids due to their high viscosity and/or viscoelasticity. The lack of long-range translational order can be expressed in another way: soft material structures are characterized by numerous defects, for example lattice dislocations or disclinations in liquid crystals (discontinuities in orientational order, Section 5.4.1). These defects have a profound effect on flow behaviour.
Another feature common to the ordering of soft materials is the periodicity of the structures formed, typically in the range 1–1000 nm, which corresponds to ‘nanoscale’ ordering. Another term often employed is ‘mesoscopic’ ordering. This originates because the length-scale of the structures is intermediate between the microscopic (atomic) and macroscopic scales.
Figure 1.2 Examples of ordering in soft materials. A nematic liquid crystal has no long-range translational order, but the molecules (here shown as ellipses) are orientationally ordered. The lamellar phase has one-dimensional translational order, the hexagonal phase two-dimensional translational order and cubic phases threedimensional orientational order. The layers in the lamellar phase can be formed from molecules (smectic phase) or amphiphilic bilayers. A hexagonal structure can be formed by disc-like molecules (then being termed columnar phase) or rod-like micelles. The cubic phase shown here is formed by spherical micelles. Bicontinuous cubic structures are also found (see Fig. 4.25d)
The number of symmetry groups of possible mesophases is restricted. Many types of soft material form structures of the same symmetry, although the molecular ordering may differ. The nematic phase (Section 5.2.2) possesses no long-range translational order, i.e. the molecular order is locally liquid-like. However, there is long-range orientational order (Fig. 1.2). Nematic phases are formed by particles ranging from small organic molecules (~2–3nm long), such as those used in liquid crystal displays, up to long macromolecules, such as rod-like tobacco mosaic virus (~300 nm long).
The simplest structure with translational order is a one-dimensional layered structure (Fig. 1.2). In thermotropic liquid crystals, there are a number of such smectic phases formed by molecules in a weakly layered arrangement (Section 5.2.2). Amphiphiles also form smectic phases, but they are usually called lamellar phases. Here, amphiphile bilayers alternate with layers of solvent (Section 4.10.2). Smectic layered structures are also found in clays, i.e. mineral-based colloidal dispersions.
Phases with two-dimensional translational order are found for both thermotropic and lyotropic liquid crystals, being termed columnar phases for thermotropic liquid crystals and hexagonal phases for lyotropic materials (Fig. 1.2). There is only partial orientational and translational order of molecules within these aggregates. This lower level of molecular order produces a ‘softer’ structure than for hexagonal phases formed by simple molecules or atoms. The hexagonal micellar phase formed by amphiphiles in solution is much softer than graphite, the structure of which is based on a hexagonal arrangement of covalently bonded atoms. The forces between molecules within rod-like micelles and those between amphiphilic molecules and solvent molecules are both much weaker than those involved in covalent bonding and the structure is much less tightly held together. In addition, the van der Waals forces act over a longer range, so that the structural periodicity is much larger. Hexagonal phases are also formed in concentrated solutions of biological macromolecules such as DNA (Section 6.3.1).
Structures with three-dimensional translational order include micellar cubic phases (Fig. 1.2) and bicontinuous cubic phases. These are distinguished topologically (Section 4.9). Micelles are discrete, closed objects within a matrix of solvent. In bicontinuous structures, space is divided into two continuous labyrinths. For lyotropic liquid crystal phases, cocontinuous water channels are divided from each other by a surfactant membrane in one type of bicontinuous structure (in the other, the positions of water and surfactant are reversed). These cubic phases are, on symmetry grounds, crystalline solids. However, unlike atomic or molecular crystals, they are built from supermolecular aggregates, i.e. micelles or surfactant membranes. Thus, as for hexagonal structures, these phases are softer than their atomic/molecular analogues, which are held together by shorter-range forces. Structures with one-, two- and three-dimensional order are also formed by block copolymers, which are polymers formed by linking two or more chemically distinct chains (Section 2.11).
The difference between direct ordering of molecules and the ‘indirect’ ordering of molecules via supermolecular aggregates is one of the distinctions between different types of soft material. Thermotropic liquid crystal phases result from partial orientational and translational order of the molecules. In contrast, the symmetry of lamellar, micellar and bicontinuous phases is specified by the location of supermolecular aggregates. The molecules within the aggregate do not have the same orientational and translational order as the mesoscopic structure itself; in fact, they can be relatively ‘disordered’.
Figure 1.3 Brownian motion of a colloidal particle results from molecular collisions, leading to a path that is a random walk. The statistical analysis for the conformation of a Gaussian polymer chain is the same (Section 2.3.2)
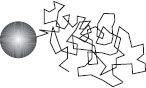
Macromolecules, colloidal particles and micelles undergo Brownian motion. This means that they are subjected to random forces from the thermal motion of the surrounding molecules. This jostling leads to a random zig-zag motion of colloidal particles, which can be described as a random walk (Fig. 1.3). Einstein analysed the statistics of a random walk and showed that the root-mean-square displacement at time t is given by
(1.5) 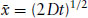
where D is the diffusion coefficient. The motion of colloidal particles in a medium gives rise to a frictional (or drag) force, which is proportional to velocity, at least if the particles are smooth and the velocity is not too great:
(1.6) 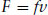
Here f is the frictional coefficient and v is the velocity. The diffusion coefficient and frictional coefficient are related to kinetic energy via
(1.7) 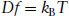
where kBT is an estimate of the translational kinetic energy per particle, kB being the Boltzmann constant. Here f for a spherical particle is given by Stokes’ law,
(1.8) 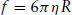
where R is called the hydrodynamic radius of the particle (i.e. the effective radius presented by the particle to the liquid flowing locally around it). Equations (1.7) and (1.8) together lead to the Stokes–Einstein equation for the diffusion of a spherical particle,
(1.9) 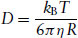
Typical diffusion coefficients for molecules in liquids (and thermotropic liquid crystals) are D ≈ 10−9 m2 s−1. Polymers are larger (i.e. they have a larger hydrodynamic radius) and so move much more sluggishly and the diffusion coefficient can be as low as D ≈ 10−18 m2 s−1. Micelles diffusing in water at room temperature with a hydrodynamic radius ≈10 nm have D ≈ 2 × 10−11 m2 s−1.
Translational diffusion of particles in the presence of a non-equilibrium concentration gradient can often be described by Fick’s first law. This states that the flux (flow), j, of material across unit area, A, is proportional to the concentration gradient:
(1.10) 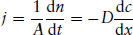
Here j is the flux, n is the amount (number of moles) of substance and dc/dx is the concentration gradient along direction x. Fick’s first law applies when the concentration gradient is constant in time. This is often, however, not the case. As diffusion occurs, the concentration gradient itself changes. Fick’s second law may then be applicable:
(1.11) 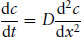
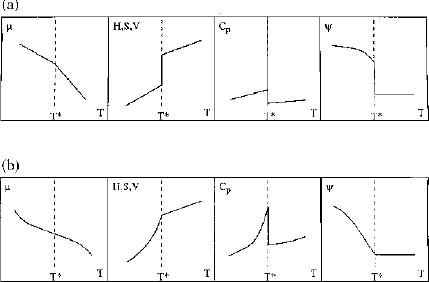
Phase transitions can be classified according to whether they are first or second order, this classification originally being introduced by Ehrenfest. Changes in various thermodynamic properties, as well as an order parameter, ψ (Section 1.6), for first- and second-order phase transitions as a function of temperature are illustrated in Fig 1.4. A first-order transition is defined by discontinuities in first derivatives of chemical potential. Enthalpy, entropy and volume can all be defined by appropriate first derivatives of chemical potential and all change discontinuously at a first-order phase transition. The heat capacity is defined as the derivative of enthalpy with respect to temperature. It is thus infinite for a first-order transition. The physical meaning of this is apparent when the boiling of water is considered. Any heat absorbed by the system will drive the transition rather than increasing the temperature, i.e. there is an infinite capacity for absorption of heat. A second-order phase transition is characterized by a continuous first derivative of chemical potential, but a discontinuous second derivative. Thus, enthalpy, entropy and volume all change continuously, although their slopes are different above and below the transition (Fig. 1.4). The heat capacity associated with a second-order phase transition is therefore finite.
Figure 1.4 Variation of thermodynamic quantities and order parameter with temperature for (a) a first-order phase transition and, (b) a second-order phase transition. The notation is as follows: μ, chemical potential; H, enthalpy; S, entropy; V, volume; Cp, heat capacity (at constant pressure); ψ, order parameter. The phase transition occurs at a temperature T = T*
Phase transitions are defined thermodynamically. However, to model them, we must turn to theories that describe the ordering in the system. This is often done approximately, using the average order parameter (here we assume one will suffice to describe the transition) within a so-called mean field theory. The choice of appropriate order parameter is discussed in the next section. The order parameter for a system is a function of the thermodynamic state of the system (often temperature alone is varied) and is uniform throughout the system and, at equilibrium, is not time dependent. A mean field theory is the simplest approximate model for the dependence of the order parameter on temperature within a phase, as well as for the change in order parameter and thermodynamic properties at a phase transition. Mean field theories date back to when van der Waals introduced his equation of state for the liquid–gas transition.
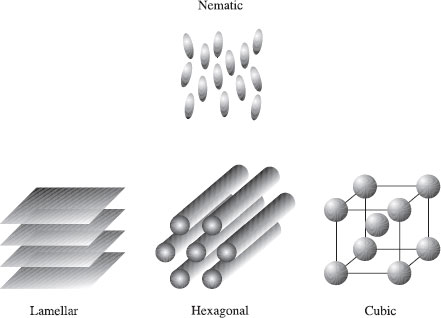
Figure 1.5 In a molecular mean field theory, one molecule (here dark) is assumed to interact with all the others through an average field (symbolized here by grey contours)

In this section we consider a general model that has broad applicability to phase transitions in soft materials: the Landau theory, which is based on an expansion of the free energy in a power series of an order parameter. The Landau theory describes the ordering at the mesoscopic, not molecular, level. Molecular mean field theories include the Maier–Saupe model, discussed in detail in Section 5.5.2. This describes the orientation of an arbitrary molecule surrounded by all others (Fig. 1.5), which set up an average anisotropic interaction potential, which is the mean field in this case. In polymer physics, the Flory–Huggins theory is a powerful mean field model for a polymer-solvent or polymer–polymer mixture. It is outlined in Section 2.5.6.
The Landau theory applies to ‘weak’ phase transitions, i.e. to continuous phase transitions or to weakly first-order transitions, where the enthalpy/entropy change is small. The order parameter is thus assumed to be ‘small’. It is a characteristic of soft materials that phase transitions are often weak. It should be mentioned in passing that the Landau theory has been applied to phase transitions in other systems, such as magnets and superconductors. However, here we consider it in the context of a phase transition in a soft material forming a low symmetry phase at low temperature and a high symmetry phase at high temperature. Such a transition is characterized by a change in an appropriate order parameter, denoted ψ, examples of which include an orientational order parameter for nematic liquid crystals (Section 5.5.1) or the composition of a diblock copolymer (Section 2.11). As shown in Fig. 1.4, for a first-order phase transition, the order parameter changes discontinuously, whereas for a second-order phase transition it decreases continuously at the transition. The Landau theory considers changes in free energy density (i.e. Gibbs energy per unit volume) at the phase transition. The essential idea of the theory is that under these conditions, the free energy density can be expanded as a power series in the order parameter:
(1.12) 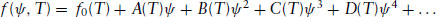
Here f0(T) is the free energy of the high-temperature phase, with respect to which the free energy is defined. The symmetry of the phases under consideration imposes constraints on the number of non-zero terms in this expansion, as illustrated by the following examples. It should be noted that ψ here is associated with a particular state of the system, the equilibrium value 〈ψ〉 being defined by the minimum of the free energy.
We now consider a second-order phase transition. Simple examples include the smectic C–smectic A transition, which is characterized by a continuous decrease in an order parameter describing molecular tilt. The smectic A (lamellar)-isotropic transition can also be second order under certain conditions. In this case symmetry means that terms with odd powers of ψ are zero. To see this consider the smectic C–smectic A transition. The appropriate order parameter is given by Eq. (5.23) and is a complex quantity. However, the free energy density must be real; thus only even terms ψψ* and (ψψ*)2, etc., remain. Then the free energy can be written as
(1.13) 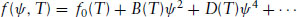
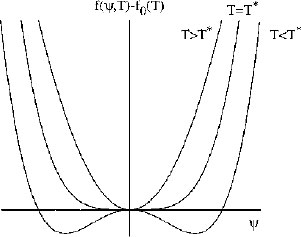
Truncation of this expansion at the fourth-order term does not lead to a loss of generality in the essential physics describing a phase transition. Typical curves according to the power series Eq. (1.13) are plotted in Fig. 1.6 above the transition temperature, T*, the free energy curve has a single minimum at ψ = 0. However, below T*, minima in free energy occur for non-zero values of ψ, as expected (Fig. 1.6). These curves are obtained with B(T) positive above the transition, but negative below the transition. At the transition B(T = T*) must vanish, and the simplest function to satisfy these conditions is
(1.14) 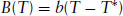
In addition, the coefficient of the quartic term should be positive, in order to obtain a stable phase below the transition. Although D(T) may vary with temperature, it is usually assumed that this dependence is weak, so that D(T) can be taken to be a constant, D. We also assume that f0(T) is not strongly temperature dependent in the vicinity of the transition. Then the temperature dependence of the free energy is determined only by B(T).
Figure 1.6 Curves of free energy with respect to that of the high-temperature phase, f(ψ, T) − f0(T), as a function of order parameter ψ, plotted according to the Landau theory for second-order phase transitions. The symmetry-breaking phase transition on reducing temperature is signalled by the development of a non-zero order parameter. At T > T*, the equilibrium value 〈ψ〉 = 0. T = T* defines the phase transition. At T < T*, the equilibrium value 〈ψ〉 ≠ 0
To find the equilibrium states, the minima in the free energy are located by differentiating it with respect to the order parameter and setting this equal to zero. The resulting cubic equation
(1.15) 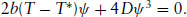
has solutions
(1.16) 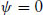
and
(1.17) 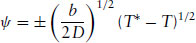
Above T*, the only real solution is ψ = 0. This corresponds to the state with an equilibrium value 〈ψ〉 = 0. However, for temperatures below T*, ψ = 0 corresponds to a maximum in the free energy (see Fig. 1.6) while the solutions ψ = ±(b/2D)1/2(T* − T)1/2 are the symmetrically placed minima. The magnitude of the equilibrium order parameter in the low-temperature phase therefore decreases with increasing temperature according to
(1.18) 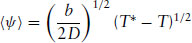
The exponent  for the temperature dependence is characteristic of mean field behaviour.
for the temperature dependence is characteristic of mean field behaviour.
The second-order nature of the transition is confirmed since the entropy change at the transition is zero. This can be shown by calculating the entropy density (at constant volume), s:
(1.19) 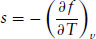
Above the phase transition this takes the value
(1.20) 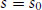
whereas below the transition,
(1.21) 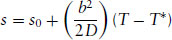
This shows that the entropy density decreases continuously to zero as the phase transition is approached, i.e. there is no discontinuity in entropy.
A similar analysis can be made for the Landau free energy of a weakly first-order phase transition, for example the nematic-isotropic transition exhibited by some liquid crystals (Section 5.7.1). The free energy (Eq. 1.13), is supplemented by an additional cubic term C(T)ψ3 if the transition is first order. The first-order nature of the transition can be confirmed by calculating the entropy density change at the transition, which turns out to be
(1.22) 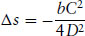
i.e. it is finite. Confirmation of this result is left as an exercise for the interested reader.
Mean field theory provides a basis for understanding many soft materials. The Landau theory is one example of a mean field theory that we have highlighted because of its generality in describing, at least qualitatively, phase transitions in weakly ordered systems. Other types of mean field theory have been used to model specific soft materials, but we do not have the space here to list them all.
Going beyond mean field models, the next level of theoretical approximation is to allow for (usually thermal) fluctuations around the mean system configuration. These fluctuations are considered to be a weak ‘perturbation’ with respect to the average order. Unfortunately, many soft materials are strongly fluctuating, especially near to a phase transition, and this approach then breaks down. This is not too much of a concern, however, because just as for atomic systems, more sophisticated perturbation theories are sure to be developed for soft materials.
Phase transitions in condensed phases are characterized by symmetry changes, i.e. by transformations in orientational and translational ordering in the system. Many soft materials form a disordered (isotropic) phase at high temperatures but adopt ordered structures, with different degrees of translational and orientational order, at low temperatures. The transition from the isotropic phase to ordered phase is said to be a symmetry breaking transition, because the symmetry of the isotropic phase (with full rotational and translational symmetry) is broken at low temperatures. Examples of symmetry breaking transitions include the isotropic–nematic phase transition in liquid crystals (Section 5.5.2) and the isotropic–lamellar phase transition observed for amphiphiles (Section 4.10.2) or block copolymers (Section 2.11).
Symmetry breaking phase transitions are characterized by a change in one or more order parameters that describe the average order in the system. In general, we can define an order parameter at a point r in the system as
(1.23) 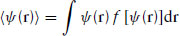
where f(ψ) is the appropriate distribution function for an orientational or translational variable.
For the isotropic–nematic phase transition, the appropriate order parameter quantifies the average degree of orientational order in the low-temperature phase. It is denoted  , as defined by Eq. (5.11) (this order parameter only quantifies the ‘first moment’ of the orientational distribution function; higher-order parameters can also be defined, as discussed in Section 5.5.1).
, as defined by Eq. (5.11) (this order parameter only quantifies the ‘first moment’ of the orientational distribution function; higher-order parameters can also be defined, as discussed in Section 5.5.1).
A phase transition from an isotropic phase to a lamellar (smectic) phase is defined by the development of translational order in one dimension. The layered structure is characterized by a periodic alternation in density or composition. An order parameter that accounts for both the phase and amplitude of the layer order is defined by Eq. (5.23). This order parameter is a vector, because the layer normals lie along a particular direction in three-dimensional space. Order parameters for phase transitions where there is a change in translational order only are vectors, whereas those associated with changes in orientational order alone are generally tensors. The orientational ordering tensor is a matrix that relates the orientation of a molecule-fixed axis system to a laboratory-fixed coordinate system (Eq. 5.5). The order parameter that is itself a scalar is equivalent to one element of the orientational ordering tensor, as discussed in Section 5.5.1.
Soft matter is characterized by complexity, both in structure and dynamics. Theories for these materials are thus difficult, and in many cases not quantitative. However, it is still instructive to consider how one variable depends on another, holding other quantities constant. This leads to so-called scaling laws, where numerical constants are omitted, but the interrelationship of quantities is established. Sometimes scaling laws can simply be deduced from dimensional arguments, i.e. by balancing the dimensions of quantities on both sides of an equation. Scaling relationships are important in the physics of soft matter, and many universal relationships can be established using them. Examples of scaling laws are given for polymer solutions in Section 2.5.1. Another example is provided by much of the discussion of intermolecular interactions in Section 1.2, where the dependence of inter-molecular potential on intermolecular separation was expressed as a power law, but without specifying an equation with precise numerical constants. A simple scaling law might look something like
(1.24) 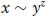
which should be read as x ‘scales with’ y to the power z. In estimating numerical quantities, the symbol ~ reads ‘of the order of’ and in this book is interpreted to mean roughly within an order of magnitude.
Unlike atoms and molecules in hard solids, colloidal particles, polymer chains and micelles are all characterized by a distribution of sizes known as polydispersity. Polydispersity has important consequences for the structure and dynamics of soft materials. It can influence phase behaviour; for example polydispersity of polymer chain length (Section 2.4.1) leads to phase separation in solutions or blends into phases rich in the small and large polymer species. Indeed, this is the basis of fractionation methods used to determine the distribution of polymer molar masses. Here a non-solvent is added to a dilute polymer solution until phase separation occurs. The critical temperature for phase separation will be reached first for large polymer chains, which can then be separated from the shorter chains remaining in solution. Another case that illustrates the effect of polydispersity on thermodynamics is provided by the crystallization of colloidal latex particles at large volume fractions (Section 3.15). This can be suppressed if the particles are polydisperse, an effect that has been modelled using binary mixtures of particles with different sizes.