
Table of Contents
Related Titles
Title Page
Copyright
Preface
1: The SNAr Reactions: Mechanistic Aspects
1.1 Introduction
1.2 Activation of the Aromatic System: Driving Force for SNAr Reactions
1.3 Leaving Group, Nucleophile, Solvent, and Medium Effects
1.4 Effects of Specific Structural Variations in the Activated Ring
1.5 Spectral Evidence for the Intermediacy of σ-Complexes in SNAr Reactions
1.6 Base Catalysis in SNAr Reactions
1.7 Regioselectivity in SNAr Reactions
1.8 Asymmetric SNAr Substitutions
1.9 Concerted SNAr Substitutions
1.10 Conclusion
References
2: Structure and Reactivity of Anionic σ-Complexes
2.1 Introduction
2.2 Structural Features of σ-Complexes
2.3 Thermodynamics and Kinetics of σ-Complex Formation
References
3: The Superelectrophilic Dimension in SNAr and Related σ-Complexation Processes
3.1 Introduction
3.2 The Classical Domain of SNAr and Anionic σ-Complexation Reactivity
3.3 Reaching the Superelectrophilic Dimension
3.4 The Synthetic Potential of σ-Complexation and SNAr Reactivity in the Superelectrophilic Dimension
3.5 Origin of the Superelectrophilicity of Neutral 10 Heteroaromatics
References
4: Synthetic Aspects of Intermolecular SNAr Reactions
4.1 Introduction
4.2 Intermolecular Displacements of a Nitro Group
4.3 Intermolecular Displacements of Halogen and Other Leaving Groups
4.4 Conclusion
References
5: Intramolecular SNAr Reactions
5.1 Introduction
5.2 SNAr Cyclizations
5.3 Smiles Rearrangements
5.4 Conclusion
References
6: Nucleophilic Aromatic Substitutions of Hydrogen
6.1 Introduction
6.2 Reactions Involving Oxidation of σ-Complex-Type Intermediates
6.3 Vicarious Nucleophilic Aromatic Substitutions of Hydrogen (VNS)
6.4 Deoxygenative SNArH Substitutions
6.5 Cine and Tele Substitutions
6.6 Conclusion
References
7: Other SNAr Substitution Pathways
7.1 SN(ANRORC) Substitutions
7.2 Radical Nucleophilic Aromatic Substitutions
7.3 Nucleophilic Aromatic Photosubstitutions
References
Index
Related Titles
Hanessian, S., Giroux, S., Merner, B. L.
Design and Strategy in Organic Synthesis
From the Chiron Approach to Catalysis
2013
ISBN Hardcover: 978-3-527-33391-2
ISBN Softcover: 978-3-527-31964-0
Nicolaou, K. C., Chen, J. S.
Classics in Total Synthesis III
Further Targets, Strategies, Methods
2011
ISBN Hardcover: 978-3-527-32958-8
ISBN Softcover: 978-3-527-32957-1
Dalko, P. I. (ed.)
Comprehensive Enantioselective Organocatalysis
2013
ISBN Hardcover: 978-3-527-33236-6
Gleiter, R., Haberhauer, G.
Aromaticity and Other Conjugation Effects
2012
ISBN Hardcover: 978-3-527-32946-5
Albini, A., Fagnoni, M. (eds.)
Handbook of Synthetic Photochemistry
2010
ISBN Hardcover: 978-3-527-32391-3
Likhtenshtein, G.
Stilbenes
Applications in Chemistry, Life Sciences and Materials Science
2010
ISBN Hardcover: 978-3-527-32388-3
Bandini, M., Umani-Ronchi, A. (eds.)
Catalytic Asymmetric Friedel-Crafts Alkylations
2009
ISBN Hardcover: 978-3-527-32380-7

The Author
Prof. Fran ois Terrier
ois Terrier
Institut Lavoisier
University of Versailles
45, Avenue des Etats-Unis
78035 Versailles, Cedex
France
All books published by Wiley-VCH are carefully produced. Nevertheless, authors, editors, and publisher do not warrant the information contained in these books, including this book, to be free of errors. Readers are advised to keep in mind that statements, data, illustrations, procedural details or other items may inadvertently be inaccurate.
Library of Congress Card No.: applied for
British Library Cataloguing-in-Publication Data
A catalogue record for this book is available from the British Library.
Bibliographic information published by the Deutsche Nationalbibliothek
The Deutsche Nationalbibliothek lists this publication in the Deutsche Nationalbibliografie; detailed bibliographic data are available on the Internet at <http://dnb.d-nb.de>.
© 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Boschstr. 12, 69469 Weinheim, Germany
All rights reserved (including those of translation into other languages). No part of this book may be reproduced in any form — by photoprinting, microfilm, or any other means — nor transmitted or translated into a machine language without written permission from the publishers. Registered names, trademarks, etc. used in this book, even when not specifically marked as such, are not to be considered unprotected by law.
Print ISBN: 978-3-527-31861-2
ePDF ISBN: 978-3-527-65617-2
ePub ISBN: 978-3-527-65616-5
mobi ISBN: 978-3-527-65615-8
oBook ISBN: 978-3-527-65614-1
Cover Design Grafik-Design Schulz, Fußgönheim
Typesetting Laserwords Private Limited, Chennai, India
Preface
It is now well documented that nucleophilic aromatic substitutions represent a class of reactions of fundamental importance in organic synthesis. In fact, the interest in the field goes back to 60 years ago when J.F. Bunnett published an authoritative review emphasizing the overwhelming importance of the presence of electron-withdrawing substituents, especially a nitro group, to achieve such reactions (Chem. Rev. 1951). This induced a number of investigations but, at the time, the focus was essentially on those substitutions that proceeded via the simple two-step addition–elimination mechanism proposed by Bunnett, known as the SNAr mechanism. As the first significant recognition of the potential of these reactions, J. Miller, one of the pioneering contributors to the field, published a book in 1968. For the most part, this book was based on the mechanistic understanding of simple substitutions and it has served as an SNAr “Bible” for 20 years.
On the basis of the information so far accumulated, the period 1970–1990 has seen many developments in the SNAr field. In addition to an improved understanding of the factors governing the two-step addition–elimination pathway formulated by Bunnett, many investigations have focused on the synthetic applicability of SNAr processes, especially those involving arene structures activated by at least one NO2 group. In this context, new reaction pathways have been discovered, which have considerably broadened the scope and importance of nucleophilic aromatic substitutions. Of particular importance was the recognition by Makosza (1983) of reactions in which an aromatic hydrogen atom can be replaced by a nucleophile under experimental conditions that lead overall to a formal SNAr departure of an extremely unstable hydride anion. The so-called vicarious and oxidative substitutions of hydrogen are representative of these reactions that give access to promising synthetic approaches. Another type of substitution proceeding formally as an SNAr reaction, but occurring in fact through multistep sequences involving nucleophilic addition, ring opening, and ring closure, was discovered by van der Plas in 1968. Commonly referred to as SN(ANRORC) substitutions, these processes provide a useful entry to the functionalization of aza-activated heteroaromatics. Other significant investigations carried out in the 1970–1990 period dealt with the photostimulation of SNAr substitutions (Havinga, Wubbels, Mutai) and the possible role of electron transfer in the reactions (Shein, Marquet, Scorrano). In the late 1980s, it become clear that the efforts so far devoted to the mechanistic understanding and synthetic applicability of SNAr substitutions have considerably enriched the perspectives offered by these versatile reactions. This convinced me to undertake a comprehensive coverage of the different aspects of activated nucleophilic aromatic substitutions in a monograph. This has resulted in the publication of the book, Nucleophilic Aromatic Displacement: The Influence of the Nitro Group (Wiley-VCH Verlag, Weinheim, 1991).
Two more decades have passed since this book appeared and the interest for SNAr substitutions has grown exponentially. In particular, the use of new methodologies has allowed SNAr substitutions to be achieved with moderately or even poorly activated aromatics or heteroaromatics, thereby extending the domain of SNAr reactivity to substrates containing no NO2 groups. Conversely, a considerable extension of the domain of reactivity to highly electrophilic substrates has also received much attention, leading to the concept of a superelectrophilic dimension in SNAr substitutions. Overall, the extension of the classical domain of SNAr reactivity has boosted the synthetic utility of these substitutions, both in intermolecular and intramolecular processes. The latter now play a key role in many macrocyclization strategies. At the same time, the SNAr synthetic potential has been improved by a better control of the regioselectivity of the reactions, whether they proceed at substituted or unsubstituted positions of the electrophilic partners. More recently, pioneering investigations of asymmetric substitutions have been carried out successfully, which represent a promising synthetic approach. The recent years have also required reconsideration of several long-accepted statements, notably those regarding the feasibility of concerted SNAr processes and the ambident electrophilic behavior of activated aromatics.
The above developments have so much enlarged the SNAr domain of reactivity that the question was posed whether a second edition of our 1991 monograph might be a simple way to keep the organic chemistry community informed of the most recent and significant advances achieved in the field, illustrating overall the remarkable versatility of SNAr substitutions. However, I became rapidly aware of the difficulty of incorporating all the new material in the framework adopted in 1991, due to two major constraints. The first constraint was purely editorial with the request that the text be not unreasonably extended. The second was more fundamental, dealing with the fact that all the SNAr topics selected as headings for my 1991 presentation have not progressed to the same extent in the last 20 years. Thus, new priorities have emerged in the coverage of the whole field, which called for special consideration and/or discussion. On the above grounds, it is finally a new book consisting of seven chapters that has come out. The most important advances in terms of synthetic and mechanistic novelty in the SNAr field are discussed in priority in six chapters. Chapter 1 deals with a mechanistic analysis of the factors governing the feasibility of SNAr substitutions, providing useful information to predict the regio- and stereoselectivity control of the reactions and to define the conditions for concerted SNAr processes. Reflecting the key role played by these species as intermediates in most SNAr reactions, Chapter 2 discusses the chemistry of anionic σ-complexes. Chapter 3 describes the new concept of superelectrophilicity, which has emerged from the kinetic and thermodynamic data collected in Chapters 1 and 2. The numerous synthetic applications of the reactions are considered in depth in Chapters 4 (intermolecular substitutions), 5 (intramolecular substitutions), and 6 (nucleophilic aromatic substitutions of hydrogen). At this stage, we were left with too little space for an exhaustive coverage of SNAr photosubstitutions, radical substitutions, and ANRORC substitutions. To cope with the objective to present a monograph involving all recognized SNAr pathways, efforts have been made to elaborate concise but, I hope, comprehensive discussions of each of these three topics, referring as much as possible to recent work. These discussions are grouped in Chapter 7.
To sum up, in this book, I have sought to provide an updated overview of SNAr substitutions, both from mechanistic and synthetic viewpoints. In so doing, I hope to stimulate further investigations in this continuously expanding field. That this book reflects some of my own interests in selecting the topics and their presentation was unavoidable in view of the large amount of work to be covered. Thus, I accept full responsibility for any significant omission in covering the literature. I hope, however, that my efforts to give a balanced presentation of mechanistic and synthetic features will be useful to workers sharing my interests in the field and to colleagues and research students who may need access to a classified but well documented review of the entire subject area. The literature has been searched approximately up to November 2012, although a few more references could be included at the proofs stage.
I wish to express my gratitude to colleagues who have read and commented on my manuscript. In particular, Erwin Buncel, François Couty, Olivier David, Julian Dust, Régis Goumont, Sergei Kurbatov, Sami Lakhdar, Herbert Mayr, and Dominique Vichard kindly read several chapters and their comments are highly appreciated. I wish also to acknowledge fruitful discussions and encouragements from a number of colleagues who share my interest for the field, in particular, Claude Bernasconi, Michael Crampton, Luciano Forlani, Jean-Claude Hallé, Mieczyslaw Makosza, and Jean-Louis Montero. Also, I am grateful to Professor Yamataka and Doctor Jean-Yves Winum for providing access to some Asian literature. My gratitude extends to former research coworkers who collaborated with me for many years and have helped so much to enhance our interest in the field, notably, Alain-Pierre Chatrousse, Taoufik Boubaker, Elyane Kizilian, Malika Mokhtari, Nizar El Guesmi, and Pedro Rodriguez Dafonte. Individual references to their published contributions are made throughout the book. I express special thanks to Karen Wright who has accepted to improve as much as possible the English text and to Anne-Marie Gonçalves and Jerôme Marrot for their assistance in drawing some structures and graphs. I am equally indebted to the Wiley staff in Weinheim, with a special tribute to Anne Brennfuehrer and Heike Noethe for their helpfulness, consideration, and patience at all stages of publication.
Finally, I wish to thank my family for their invaluable support during the writing of this book.
Versailles, April 2013
Francois Terrier
1
The SNAr Reactions: Mechanistic Aspects
A general nucleophilic aromatic SNAr substitution can be described by Eq. (1.1), in which Nu represents an anionic or a neutral nucleophile and L a good leaving group or nucleofuge. A leaving group L can bear no charge (F, Cl, Br, I, NO2, OR, OSO2R, etc.), becoming negatively charged following displacement, or it can be positively or negatively charged (NR3+, SO3−), becoming uncharged or more negatively charged when displaced [1–8]. The abbreviation EWG is used to denote the presence of one or more (electron-withdrawing group)s (e.g., NO2 in the aromatic ring). Because the presence of an EWG is of fundamental importance to induce the process, an SNAr reaction is commonly referred to as an activated aromatic substitution process. Interestingly, Eq. (1.1) also applies to many heteroaromatic systems, whether they exhibit some intrinsic π-deficiency, for example, pyridine and other fully aromatic nitrogen heterocycles, or not, for example, pyrroles, thiophenes, and furans [9].
1.1 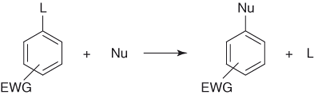
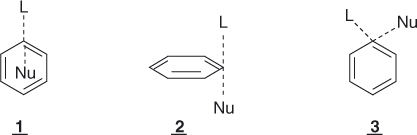
Major features of SNAr substitutions are that they occur without rearrangement and, when uncatalyzed, they display kinetics and response to structural and environmental factors that indicate a bimolecular mechanism [1–8]. In this regard, SNAr processes are formally similar to aliphatic nucleophilic SN2 substitutions but the view that the two displacements could proceed along analogous reaction paths was rejected quite early [1–3]. Should aromatic substitutions go via a concerted mechanism of the type established for SN2 reactions (Eq. (1.2)) would imply the formulation of transition-state models in which the benzene resonance is retained (e.g., 1, 2, or 3). However, all models of this sort were discarded by Bunnett and Zahler [1] because of violation of the Pauli principle and/or inconsistency with spatial requirements.
1.2 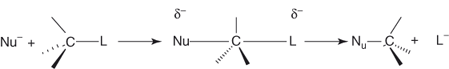
Therefore, an addition–elimination mechanism has been postulated by Bunnett for SNAr processes [1]. As a first step, this involves addition of the nucleophile to the aromatic electrophile to form an intermediate cyclohexadienyl anion of some stability in which the carbon center undergoing the substitution becomes sp3-hybridized – that is, the benzenoid resonance is broken. This intermediate, also known as a σ-complex intermediate, subsequently decomposes to give the substitution product. For anionic nucleophiles, the process is outlined in Eq. (1.3) and illustrated by the potential energy diagrams of Figure 1.1, which show that, depending on the relative energies of the two transition states, either the formation or the decomposition of the anionic intermediate 4 may be rate limiting. Reasonable structures for these transition states are 5 and 6, respectively [1–6].
1.3 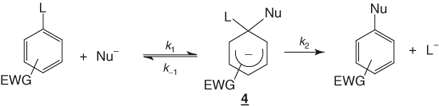
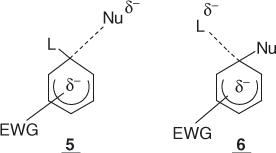
Figure 1.1 Energy diagrams for SNAr reactions of Eqs. (1.3) and (1.4) (a) assuming rate-limiting formation or (b) rate-limiting decomposition of the intermediates 4 or 7.
For neutral nucleophiles (e.g., water, alcohols, amines), the postulated mechanism is shown in Eq. (1.4). In this case, the initially formed σ-adduct 7 is zwitterionic and in most cases contains an acidic proton, which can be removed by a base such as the nucleophile itself. Conversion of 7 to products can therefore occur via an uncatalyzed k2 pathway or via a base-catalyzed  pathway. In the absence of base catalysis, energy profiles similar to those of Figure 1.1 can be envisioned for Eq. (1.4) [1–6].
pathway. In the absence of base catalysis, energy profiles similar to those of Figure 1.1 can be envisioned for Eq. (1.4) [1–6].
1.4 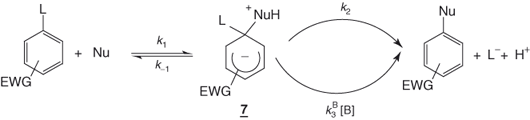
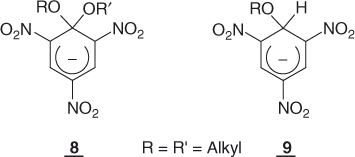
There is now ample evidence that the stepwise mechanisms depicted in Eqs. (1.3) and (1.4) fit very well the great majority of intermolecular and intramolecular nucleophilic displacements involving aromatic and heteroaromatic substrates [1–6]. Although the matter has been the subject of many reviews [1–9], the major purpose of this chapter is to provide an updated as well as comprehensive discussion of the basic features that have together contributed to the progress in our knowledge of these stepwise SNAr substitutions. Probably, the most convincing evidence is the successful and unambiguous NMR identification of intermediates of type 4 along some substitution pathways [10, 11]. This goes with the evidence gained from the structural analogy between the postulated reaction intermediates 4 and 7 and the stable σ-complexes of the type 8 and 9 identified quite early by Jackson and Meisenheimer, and described in Chapter 2 [12, 13]. Also of fundamental importance are the results of detailed kinetic studies of a large number of substitutions. These revealed leaving group and nucleophile effects as well as typical acid–base catalysis phenomena that can be understood only in terms of the intermediacy of the adducts 4 and 7 [1–6, 14, 15].
Until the 1990s, the addition–elimination mechanisms of Eqs. (1.3) and (1.4) were considered an unquestionable feature of SNAr reactions. It was therefore a major event in 1993, when Williams and coworkers provided kinetic evidence that some SNAr substitutions may proceed preferentially through a concerted mechanism [16]. A representative example is the phenolysis of 2-(4-nitrophenoxy)-4,6-dimethoxy-1,3,5-triazine that takes place in one step via the transition state 10 (Scheme 1.1, path a) and not via the expected initial formation of the σ-adduct 11 (Scheme 1.1, path b) [16]. Following Williams' discovery, a number of theoretical studies of SNAr reactions have been carried out, which have provided pertinent information showing that concerted processes remain the exception [17–20]. Accordingly, for clarity, the primary focus of this chapter is the understanding and feasibility of the stepwise substitutions, as formulated in the general Eqs. (1.3) and (1.4). The possibility of concerted SNAr substitutions is addressed in a separate discussion in Section 1.9.
Scheme 1.1 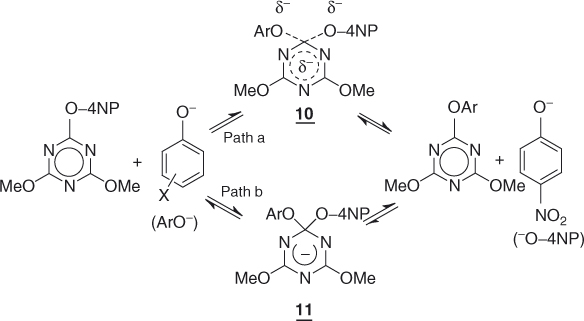
Some kinetic and theoretical studies have supported the idea that noncovalent bonding may take place initially between the electrophile and nucleophile partners, resulting in the formation of molecular complexes – referred to as electron donor–acceptor (EDA), charge-transfer (CT), or π-complexes – which in some cases could be detectable [21–27]. This question is considered only when needed in this chapter but a detailed coverage of these interactions is out of the scope of this book.
Because of the evident repulsion between a π-electron system and an approaching nucleophile, nonelectron-deficient benzene derivatives are intrinsically reluctant to suffer nucleophilic addition. On these grounds alone, it can be understood why the presence of EWGs is a key factor that determines the feasibility of SNAr reactions in general (Eq. (1.1)). Introduction of substituents such as NO2, CN, COR, CF3, and SO2R has the effect of lowering the electron density of the benzenoid system, especially at the o- and p-carbons, thus favoring nucleophilic attack at these positions. It remains therefore to be ascertained whether the process can result in the formation of relatively stable cyclohexadienyl intermediates such as 4 (Eq. (1.3)) and 7 (Eq. (1.4)).
Let us consider first the unsubstituted cyclohexadienyl anion 12 and the contributing resonance structures 12a, 12b, and 12c. These structures do not violate any quantum-mechanical principle, while suggesting that the cyclohexadienyl anion can retain an appreciable amount of the resonance energy of the parent aromatic ring. More important, it could be anticipated that such structures will be more favored if the negative charge can be dispersed through electron-withdrawing substituents, especially those that are capable of conjugation in the positions ortho and para to the sp3 carbon. In particular, introduction of a group exerting a strong −M-effect, such as NO2, in these positions should lead to significant stabilization of 12, as shown in structures 13a ↔ 13b and 14a ↔ 14b.
1.5 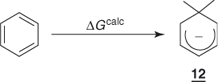
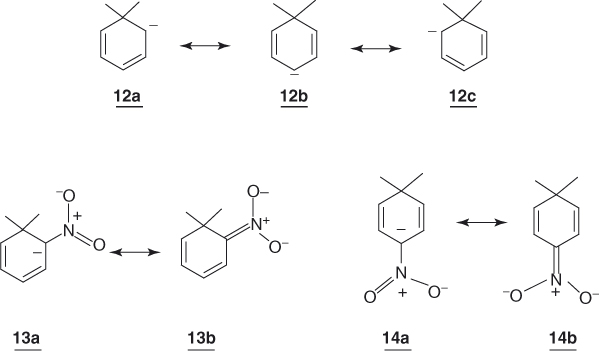
These predictions agree with theoretical calculations as well as with experimental data. Dewar [28] has calculated that formation of a cyclohexadienyl ring from a benzene ring is associated with a decrease of only 41.80 kJ mol−1 (ΔGcalc in Eq. (1.5)) in resonance energy; that is, 12 retains a large portion of the original stabilization energy. On the other hand, a number of calculations have been made that have confirmed that 12 may be properly represented by valence structures 12a ↔ 12b ↔ 12c and have indicated a greatest π-population at the para position [29–31]. Analysis of 1H and 13C NMR data obtained in liquid ammonia for the cyclohexadienyl anions 15, which were produced by proton abstraction from the corresponding 1,3-dienes or 1,4-dienes, have led to similar conclusions [30]. Importantly, these studies have also demonstrated that the ring system of such anions is planar, with no homoaromatic overlap1 occurring [29–31, 33].
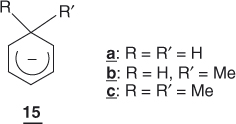
1.6 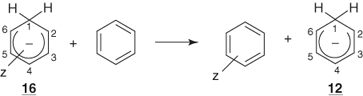
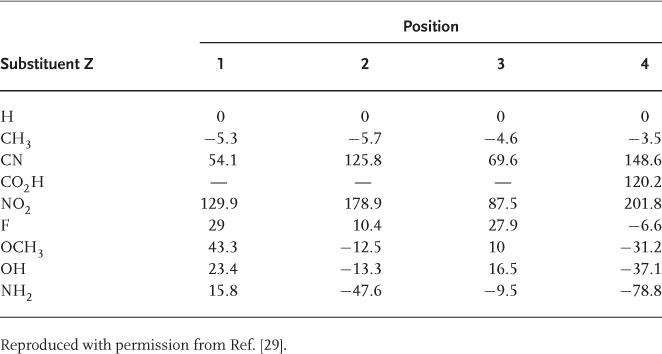
Calculations of the energies of various monosubstituted cyclohexadienyl anions 16 have been made and used to derive the stabilization energies of these anions relative to substituted benzenes [29]. These energies were defined as the energy changes for exchange reactions of the type shown in Eq. (1.6). Some data are presented in Table 1.1, in which a positive value indicates a greater stability of the substituted anion (16) compared to the unsubstituted anion (12). As can be seen, all EWGs exert a stabilizing effect, but the calculated stabilization energies are much larger for NO2 than for other common groups, for example, CN or CO2H, in full accord with the early recognition of the preeminent role of the NO2 substituent in determining the feasibility of SNAr substitutions [1–6]. Importantly, the calculations emphasize two other features: (i) the stabilization increases in the order meta  ortho < para, confirming the primary role that resonance structures, for example, 13b and 14b for a NO2 substitution, must play in determining the stabilities of cyclohexadienyl anions. In contrast, most electron-donor substituents, for example, F, OCH3, OH, and NH2, exert a destabilizing effect in the ortho and para positions [29] and (ii) there is a substantial stabilization of the cyclohexadienyl structure when substituents exerting a strong −I-effect are bonded at the ipso-carbon (C-1). Being especially large for NO2, this α-effect is also significant for F and OR, a result that accounts in part for the good nucleofugality of these groups in SNAr reactions (see later) [2].
ortho < para, confirming the primary role that resonance structures, for example, 13b and 14b for a NO2 substitution, must play in determining the stabilities of cyclohexadienyl anions. In contrast, most electron-donor substituents, for example, F, OCH3, OH, and NH2, exert a destabilizing effect in the ortho and para positions [29] and (ii) there is a substantial stabilization of the cyclohexadienyl structure when substituents exerting a strong −I-effect are bonded at the ipso-carbon (C-1). Being especially large for NO2, this α-effect is also significant for F and OR, a result that accounts in part for the good nucleofugality of these groups in SNAr reactions (see later) [2].
Table 1.1 Stabilization energies (kJ mol−1) of substituted cyclohexadienyl anions, as defined by Eq. (1.6).
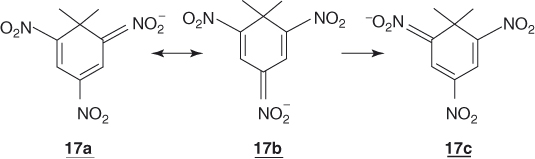
The stabilizing energy brought about by the introduction of several activating groups in 12 has not been estimated via Eq. (1.6) but other calculations have been made that leave no doubt that the introduction of additional EWG in the ortho and/or para position(s) results in an increased stabilization of the anion [4, 14, 34–37]. In the case of 2,4-dinitro- and 2,4,6-trinitro-derivatives, the calculations also predict that significantly more charge is located on the p-NO2 group than on the o-NO2 groups; that is, the resonance structure 17b is favored relative to 17a and 17c. As further elaborated in Chapter 2, this prediction is fully consistent with the experimental information derived from X-ray structure determination or NMR characterization of many stable σ-adducts.
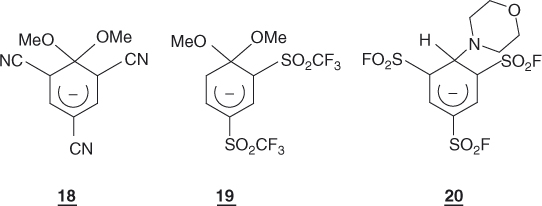
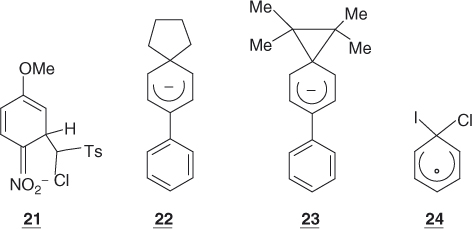
On the above theoretical grounds, it is quite reasonable to envision that a number of SNAr substitutions will proceed through the addition–elimination mechanisms of Eqs. (1.3) and (1.4), involving cyclohexadienyl anions of sufficient stability, to be true intermediates along the reaction coordinates. Interestingly, there are experimental data that are in agreement with this view. For structural reasons to be discussed later in this chapter, many activated arene–base interactions cannot proceed further than the addition step, providing opportunity for a direct study of the formation of potentially stable σ-adducts [4, 6, 13, 38]. The long-known trinitro adducts 8 and 9 are prototype examples but many other trinitrobenzene (TNB) as well as dinitrobenzene adducts have been characterized, including in the gas phase, by X-ray crystallography (Chapter 2) [39–43]. A large number of adducts derived from activated arenes containing no NO2 groups have also been identified, for example, 18–20 [44–46]. Also consistent with theoretical predictions, the experimental evidence is that the stability of the adducts strongly decreases as the number and/or efficiency of activating groups in the benzene ring is reduced, making it difficult to firmly identify monosubstituted adducts. The only unequivocal exception is the recent NMR characterization of the adduct 21 that formed initially as a mixture of two diastereomers on treatment of 4-nitroanisole with the carbanion of α-chloromethyl p-tolyl sulfone [47]. However, increasing the possibility of delocalization of the negative charge through an exocyclic phenyl ring has allowed the NMR characterization of the spiro adducts 22 and 23, which contain no EWG groups. They are formed from the reaction of biphenylylchloroalkanes with alkali metals in THF (tetrahydrofuran) at −70 °C [48]. As a remarkable example of an SNAr stepwise process in which the activation is provided by the presence of a positively charged leaving group, there is the reaction of the iodobenzene cation with Cl−. Using femto second resolution, Zewail et al. have demonstrated the formation of the intermediate σ-complex 24 – in this case, a delocalized cyclohexadienyl radical – along the reaction coordinate [49].
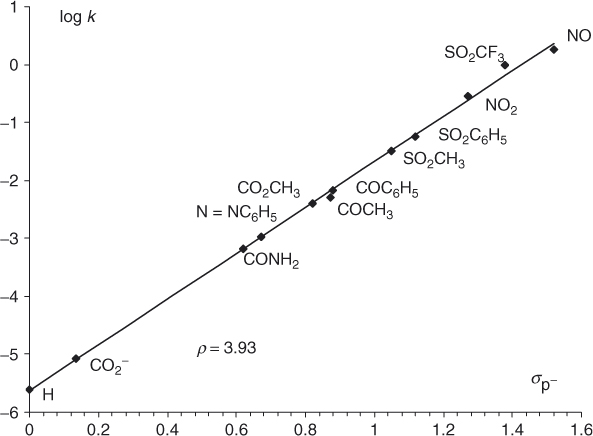
Clearly, the existence and stability order of firmly identified σ-adducts reinforces the view provided by the calculations, which refer for the most part to isolated molecules in the gas phase, that cyclohexadienyl intermediates can initially form in most SNAr reactions of activated benzenes. Should this be the case, Miller [2] has suggested that the two transition states for the addition of the nucleophile and elimination of the nucleofuge, for example, 5 and 6, are probably more similar in energy to the intermediates (i.e., 4 or 7) than to either reactants or products. On this ground, the overall rates of substitution should be reflected in the stabilities of the intermediates, and rate data for homogeneous sets of substitutions should be successfully interpreted by consideration of the effects of substituents on the stability of these intermediates [2]. In particular, the rates of substitutions involving rate-determining addition of the nucleophile should increase with increasing substituent constant. Figure 1.2 shows that such correlations have actually been observed in some systems in which the para substituent was varied [2, 50], giving real support to two-step processes. A point of importance in this reasoning is that the Hammett correlation of Figure 1.2 requires the use of the σp− constant for substituents such as NO2, CO2R, and COR, which interact conjugatively with the reaction center and thus serve to absorb the negative charge, as depicted in 14b. Figure 1.2 also reveals that except for NO and SO2CF3, NO2 is the most activating para substituent in the series of reactions at hand. The greater activating power of an SO2CF3 group (σp = 0.96, σp− = 1.63) relative to a NO2 group (σp = 0.78; σp− =1.27) in the benzene series is well substantiated, being clearly the result of a Fπ effect [13, 51–56]. Recent studies by Yagupolskii et al. have shown that CF3S(O) = NSO2CF3 (σp = 1.35; σp− = 2.90) and SO2F are superstrong electron-withdrawing substituents, notably in SNAr reactions [57–60]. Other work indicates that the positively charged N2+ group is more activating than NO2 [2].
Figure 1.2 Hammett correlation for the reactions of 1-chloro-2-nitro-4-Z-substituted-benzenes with methoxide ion in methanol at 50 °C (ρ = 3.93; k in liters per mole per second).
(Data reproduced with permission from Ref. [2]. The point for Z = SO2CF3 is taken from Ref. [50].)
With the same reasoning, the data in Table 1.1 predict that substitutions involving ortho-activated benzenes should proceed at lower rates than those involving the para isomers. Experimental data do not always conform to this prediction and there are many examples of a similar or even greater activation by an ortho substituent than by a para substituent [1, 2, 5, 6, 61]. As an example, o-nitro- and p-nitro-fluorobenzenes undergo isotopic exchange of 18F− at similar rates in DMSO [62]. Also noteworthy is the especially strong activation exerted by an o-carboxamido group in SNAr reactions involving anionic nucleophiles [63]. This occurs because the reactivity of ortho-substituted derivatives is a function of other variables such as steric factors, polarizability, field, and electrostatic effects and also hydrogen bonding (the so-called built-in solvation; see Section 1.4.1) [1, 2, 4, 6].
NO2 being the most commonly employed activating substituent in SNAr reactions, it is a significant result that the reactivity order anticipated from the additivity in the stabilization energies of the cyclohexadienyl intermediates – as listed in Table 1.1 with no consideration of other influences such as steric factors associated with the presence of ortho substituents – is in qualitative agreement with the sequence revealed by rate studies involving comparable systems. For methoxydefluorination substitutions activated by NO2 groups in methanol, we thus have the following sequence [6]:
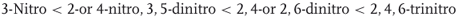
This ordering calls for two comments: (i) activation by two m-nitro groups is of the order of that provided by one o- or p-nitro group, allowing in many instances clean SNAr processes to occur under mild experimental conditions [6, 64] and (ii) despite its weak activating effect, one m-nitro group can promote synthetically important SNAr processes providing the presence of very good leaving groups and the use of the most efficient experimental conditions in terms of temperature, solvent, catalyst, and so on [6, 65]. As elaborated in more detail in Chapters 4 and 5, a judicious design of the reaction protocols allowed in fact to achieve a number of substitutions involving substrates bearing only one moderately activating substituent in the ortho/para or meta positions, for example, CN, CHO, COR, CF3, SOR, SO2R, SO2NR2, and –N=NC6H5 [66–71]. As a practical example, displacement of a fluorine leaving group activated by a ketone group at the meta position was successfully achieved in the synthesis of hyperbranched poly(etherketone) that does not require complete conversion [72]. Heterocyclic azole units exhibiting σp values of the order of those of the above substituents are also efficient in promoting SNAr substitutions, for example, the activation provided by an oxazoline moiety in Meyers methodology for the synthesis of biaryl systems (Section 1.2.4) [73].
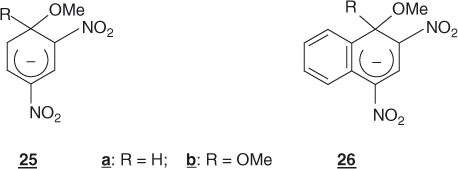
Extension of the aromatic system through benzoannelation enhances the stability of a cyclohexadienyl anion, mainly because of an increased delocalization capability of the negative charge [4, 6, 13, 74]. Thus, naphthalene σ-adducts such as 26a and 26b are thermodynamically more stable than the benzene analogs 25a and 25b. Accordingly, naphthalene derivatives are more prone to SNAr substitutions than similarly activated benzenes; for example, 1-chloro-2,4-dinitronaphthalene reacts 10–100 times faster than 1-chloro-2,4-dinitrobenzene with common nucleophiles such as CH3O− or aniline, even though there is evidence that the proximity of the peri position may sterically affect the reactions [1].
Pyridine and other fully aromatic nitrogen heterocycles – often considered to be aza-substituted arenes – are π-deficient [4, 9]. Replacement of a ring carbon in an arene system by a more electronegative nitrogen atom results in a greater electron density on that atom, with a concomitant reduction in electron density on the remaining carbon atoms. This deficiency favors nucleophilic attack, and therefore nucleophilic substitution of a suitable leaving group, especially at the α- and γ-positions to the heteroatom (resonance structures 27a–c). Concomitantly, the formation of the corresponding aza-cyclohexadienyl intermediates 28 and 29 is facilitated by the somewhat lower aromaticity of the parent substrate and the ability of the nitrogen atom to stabilize such adducts by accommodating the negative charge, as shown in 28b and 29b. Contrasting with the failure to form 12 from benzene, the aza σ-adducts 30a and 30b have been directly obtained on treatment of pyridine with butyllithium in ether or in hexane and phenyllithium in N,N,N′,N′-tetramethylethylene diamine, respectively [75–77]. Both calculations and NMR studies indicate that the ring systems of the 2-pyridyl anion 28 and the 4-pyridyl anion 29 are planar, showing that introduction of the aza functionality does not affect the geometry of the cyclohexadienyl anion [78, 79].
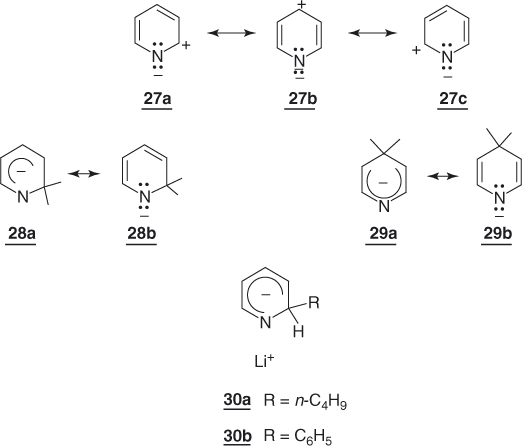
In SNAr substitutions, the activating effect of the aza group at both the 2- and 4-positions lies not far – about one order of magnitude – below that of a NO2 group [9]. The highest capacity of the NO2 group to delocalize the developing negative charge in the related transition state accounts for this frequently observed trend in reactivity [9, 14, 80, 81]. In some instances, however, solvent and hydrogen-bonding effects may favor the aza activation [9]. This is demonstrated in Table 1.2, which compares some rate data obtained for a few typical pyridine substrates with those for the benzene analogs [2, 17, 82–91]. Because of the intrinsic activation by the aza group, introduction of one or two nitro groups in the 3- and/or 5-positions yields nitropyridines or dinitropyridines that are almost as reactive as the analogous dinitro- or trinitrobenzenes [9, 14, 82, 92].
Table 1.2 The activating effect of the aza group in SNAr substitutions.
Further evidence for a strong activating effect of the aza group is given by the observation that 1,3-diazines such as 4-chloropyrimidine or 2-chloropyrimidine show an SNAr reactivity approaching that of 2-chloro-5-nitropyridine and 2-chloro-3-nitropyridine, respectively [9, 83, 84]. On the other hand, comparison of the last two entries in Table 1.2 shows that, despite the electron-donating effect of the anilino moiety, the hydrolysis of the symmetrical 2-anilino-4,6-dichloro-1,3,5-triazine proceeds essentially at the same rate as that of picryl chloride in aqueous solution [93, 85]. The notable activation of monoazines (including quinolines) of diazines (including pyridazines, pyrazines, naphthyridines) and related substrates such as purines, and of triazines (including the nonsymmetrical 1,2,4-triazines) is also demonstrated by the successful characterization of a number of σ-adducts, for example, 31–35 [94–98].
In view of the relatively similar activation provided by aza and NO2 groups, it is reasonable to anticipate that SNAr substitutions of pyridine and other azine derivatives will proceed through two-step pathways of the type shown in Eqs. (1.3) and (1.4). As a matter of fact, the observation of base catalysis in substitutions of haloazines with amine nucleophiles is consistent with Eq. (1.4) [14]. Another significant finding is the observed departure sequence F > Cl, Br > I in the SNAr substitutions of 6-X-substituted purine nucleosides with aliphatic amines as well as methoxide or thiolate anions in acetonitrile or DMSO, as depicted in Eq. (1.7) [99]. As discussed later, the sequence is the one expected for a rate-limiting nucleophilic addition step in Eqs. (1.3) and (1.4).
1.7 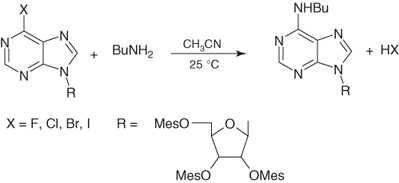
Activation at the α- and γ-positions in the pyridine ring is increased on conversion of the nitrogen atom into an N+–R or an N+–O− group, both functionalities being much more activating than NO2 [2, 86, 100, 101]. However, nitropyridines are so weakly basic (pKa < 0) that quaternization of the aza group due to protonation does not commonly occur under the experimental conditions required to achieve most SNAr substitutions of these derivatives [9]. Also to be noted is that treatment of nitropyridines, nitropyrimidines, and other azine derivatives with strong bases may result in nucleophilic substitution pathways involving ring-opening and ring-closure processes, that is, the SN(ANRORC) mechanism [3, 102]. These particular systems are considered in Chapter 7.
Five-membered ring heterocycles such as pyrrole, furan, thiophene, and selenophene possess π-excessive character. This implies low intrinsic reactivity toward nucleophilic reagents, comparable to that of benzene. Accordingly, only substrates bearing suitably located EWGs undergo facile SNAr substitutions in these compounds. In accordance with simple theory, the substitutions proceed in general more readily at the α- than at the β-position to the heteroatom [9].
The effectiveness of SNAr reactions at the α-position implies the formation of hetarenide intermediates such as X-36, where Y and Z denote EWG groups (e.g., NO2, CN, CHO, CO2R) [103]. Compared to the benzene systems, the lower aromaticity of the five-membered rings and the ability of heteroatoms such as O, S, and Se to accommodate the negative charge are two factors that favor the formation of X-36 [4, 6, 9]. Another favorable factor is that the attainment of the tetrahedral geometry at the reaction site in a σ-adduct involves much less bond strain in five-membered than in six-membered rings [6, 9, 13b, 104] In agreement with these views, theoretical calculations suggest a much greater stability (about 50 kJ mol−1) of the dinitrothiophene anion X-36 (X = S, Y = Z = NO2) than of the dinitrobenzene analog 37 [1, 6, 105].
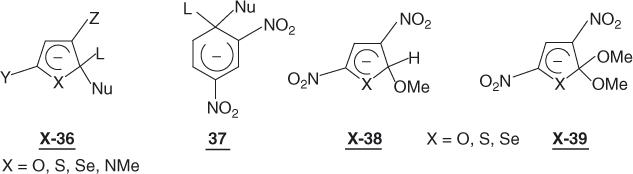
The conclusions reached by theoretical calculations are confirmed by measurements of the stabilities of the firmly characterized dinitroadducts X-38 and X-39 [13b]. For these substrates, the stability order is
in agreement with the expected influence of the heteroatom on the tendency to complexation of the heterocycles. Going from X = O to Se or S to N–R reduces the electron-withdrawing influence of the heteroatom and increases the aromatic character of the reactant molecules, thus decreasing the stability and the ease of formation of the adducts. The effect is especially important for the formation of the pyrrole adducts, which are considerably less stable than their furan, selenophene, or thiophene analogs [1, 6].
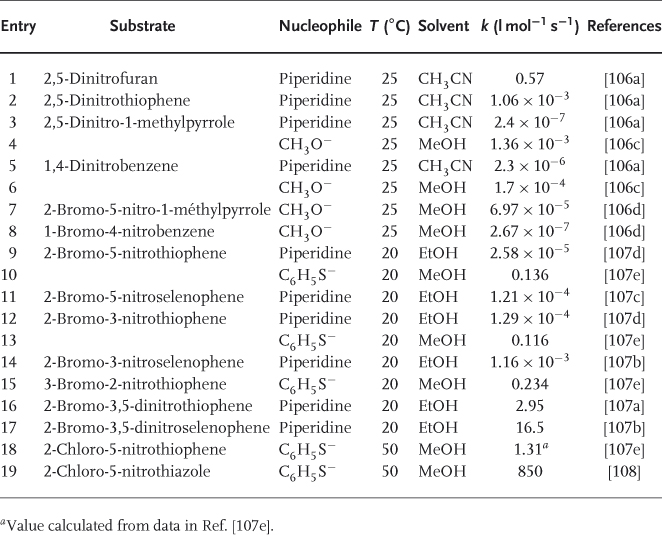
Table 1.3 shows that the SNAr reactivities of these heterocycles parallel nicely the order of stability found for the corresponding σ-adducts. Thus, the rate constants for nucleophilic displacement of a NO2 group from 2,5-dinitrofuran, 2,5-dinitrothiophene, and 2,5-dinitro-1-methylpyrrole by piperidine and p-toluenethiolate ion decrease in the order O > S  N–R [106]. Depending on the nucleophile, nitro-activated pyrroles react more rapidly or more slowly than the corresponding nitrobenzenes [106]. Also illustrated is the greater reactivity of nitroselenophenes compared to nitrothiophenes [107].
N–R [106]. Depending on the nucleophile, nitro-activated pyrroles react more rapidly or more slowly than the corresponding nitrobenzenes [106]. Also illustrated is the greater reactivity of nitroselenophenes compared to nitrothiophenes [107].
Table 1.3 Reactivity of activated five-membered ring heterocycles in SNAr reactions.
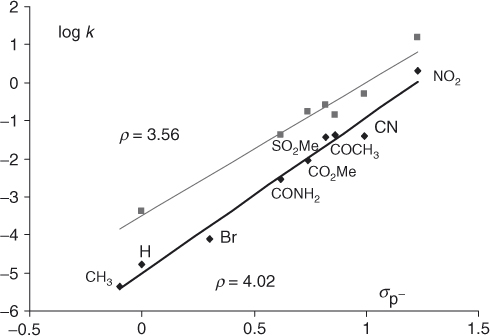
Changes in reactivity caused by variations in the nature of the activating substituents have been in many cases successfully correlated by Hammett relationships [107, 109, 110]. The kinetics of piperidino debromination of 2-bromo-3-nitro-5-Y-thiophenes and the corresponding selenophenes in ethanol have been very informative (Eq. (1.8)) [107, 109–111]. The high positive ρ values (≈3.2) found for these reactions, together with the necessity of using σp− instead of σp constants in the correlations indicate that the transfer of the negative charge has made notable progress in the transition states, suggesting a complex-like structure [109]. Importantly, the Hammett equation was also found to fit changes in reactivity brought about by variations in the nature of the ortho-like Z substituents [111]. Figure 1.3 shows the Hammett plots for substitution of various 2-bromo- and 2-p-nitrophenoxy-3-Z-5-nitrothiophenes with piperidine in methanol (Eqs. (1.9a) and (1.9b)). The existence of such linear energy ortho correlations is peculiar to five-membered ring heterocycles, where steric effects of substituents ortho to the site of the nucleophilic attack are minimized. Of further interest is that the ρ values for the reactions shown in Eqs. (1.9a) and (1.9b) (≈4.02 and 3.56, respectively) are higher than those for the reactions of Eq. (1.8), indicating that substituents exert a higher electronic effect on the reaction center in the ortho-like rather than in the para-like position. This behavior which has been termed the “hyperortho relationship,” is now well documented in the chemistry of five-membered ring heterocycles [110, 111].
Figure 1.3 Hammett correlations for the reactions of various 2-bromo- (lower line) and 2-(p-nitrophenoxy)- (upper line) -3-Z-5-nitrothiophenes with piperidine in methanol at 20 °C (see Ref. [109] for the σp-values employed in the correlations); k in liters per mole per second.
(Data reproduced with permission from Ref. [109].)
1.8 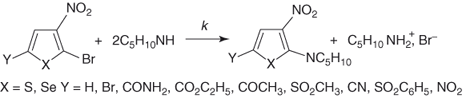
1.9 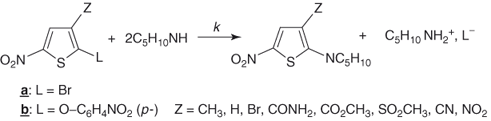
Although the susceptibility to nucleophilic attack is weaker at the β- than at the α-position to the heteroatom, identification of the stable σ-adducts 40 and 41 supports the idea that hetarenide intermediates of type 42 may form in nucleophilic substitutions of activated five-membered ring heterocycles bearing a leaving group in the 3-position. Table 1.3 compares the reactivity of 2-nitro-3-bromothiophene with that of its 3-nitro-2-bromo and 5-nitro-2-bromo isomers toward benzenethiolate in methanol.
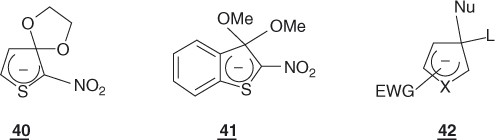
Introduction of a second electronegative heteroatom into five-membered ring systems may result in a large increase in the tendency toward nucleophilic substitution, especially if there is also activation by a suitably located EWG such as NO2 [2]. Thus, it is shown in Table 1.3 that 2-chloro-5-nitrothiazole is about 650 times more reactive than 2-chloro-5-nitrothiophene toward benzenethiolate anion [108]. Nitropyrazoles and nitroimidazoles also have a high reactivity, but the reactions are often complicated by side processes [112, 113]. It is to be noted that the versatile biological activity of nitrothiophenes is closely related to their SNAr susceptibility to react with intracellular sulfur-containing proteins. Abnormal reactivity patterns are also the rule with 3,4-dinitrothiophenes [111, 113, 114].
A number of electron-withdrawing heterocyclic functionalities exhibiting an activating effect (σp = 0.5–0.6), comparable to that of CN or CF3 groups have been successfully employed to activate SNAr displacements of good leaving groups, such as F, NO2, and OCH3, in aromatic systems [70, 115]. These include, among others, azomethine, thianthrene, benzoxazole, benzotriazole, quinoxaline, benzimidazole, oxadiazole, triazole, phenylquinoxaline, and triazine moieties located in a para or an ortho position relative to the leaving group. Many azole-activated SNAr transformations involving departure of a NO2 or F group have been described in the recent literature [70, 115, 116]. In particular, this activation has proved to be very efficient in the synthesis of a variety of polyaryl ethers incorporating the heterocyclic units [70, 115]. Another heterocyclic moiety worth mentioning, which is utilized to promote SNAr substitutions is an oxazoline ring. In fact, an oxazoline-mediated SNAr methodology developed by Meyers et al. has been extensively employed in the synthesis of biaryl systems as well as of numerous important aromatic targets (Eq. (1.10)) [73, 117].
1.10 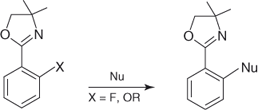
On annelation of a benzene ring by a variety of electron-withdrawing five-membered heterocyclic moieties, 10π-electron ring systems exhibiting a very high tendency toward SNAr substitution are obtained [118]. 2,1,3-Benzoxadiazoles and related 1-oxides, commonly known as benzofurazans and benzofuroxans, respectively, are representative structures. A first striking instance of the ease of SNAr substitution is found with 4-chloro-7-nitrobenzofurazan – the commercially available NBD-Cl (4-chloro-7-nitrobenzofurazan) – reacting with a variety of base reagents, such as amines or hydroxide, alkoxide, and thiolate ions [119–122]. In fact, the reactivity of this mononitro-activated substrate in Eq. (1.12) is comparable to that of picryl chloride in Eq. (1.11), with rate constants of 7.7 and 17 l mol−1 s−1 for reaction of these compounds with methoxide ion in methanol, respectively [119, 123]. Importantly, the feasibility of the SNAr process of Eq. (1.12) goes along with the successful identification of mononitro adducts of type 43 all of which have a thermodynamic stability comparable to that of TNB analogs, that is, 8 and 9 [13b]. Also revealed by Table 1.4, the rates of substitution of 7-L-4-nitrobenzofurazans and 4-L-5-nitrobenzofurazans, as well as of the related benzofuroxans, with MeO− are similar. 5-Halo-4-nitrobenzofurazans exhibit a high susceptibility to SNAr methoxydehalogenation [119, 124].
Table 1.4 Reactivity of nitrobenzofurazans and nitrobenzofuroxans.
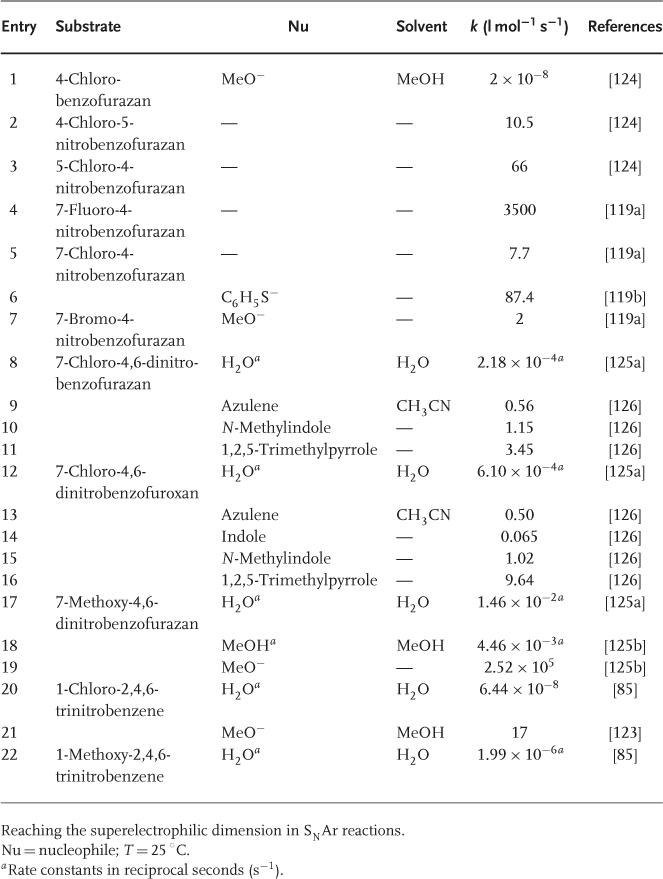
1.11 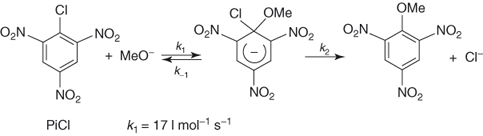
1.12 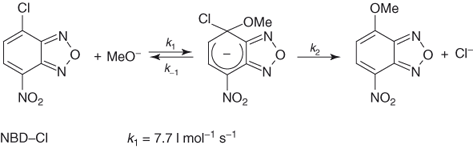
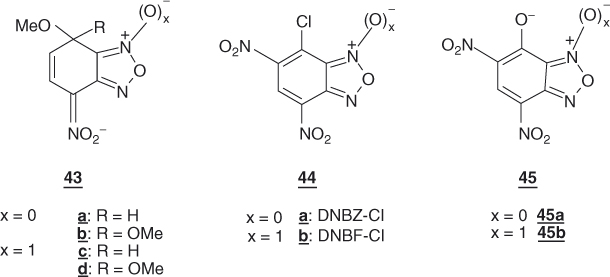
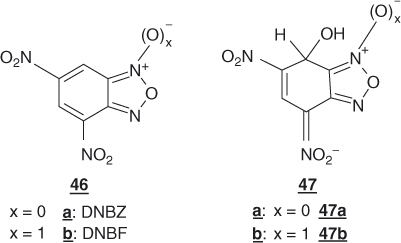
Obviously, it could be anticipated that addition of a second nitro group to NBD-Cl will project the resulting substrate, namely, 4,6-dinitro-7-chlorobenzofurazan (DNBZ-Cl; 44a) in a domain of SNAr reactivity inaccessible with aromatic or heteroaromatic structures so far considered. This expectation is fully materialized when DNBZ-Cl and the related 4,6-dinitro-7-chlorobenzofuroxan (DNBF-Cl; 44b) are allowed to react with a variety of weak or very weak nucleophiles. Thus, Table 1.4 shows that both DNBZ-Cl and DNBF-Cl hydrolyze very rapidly on reaction with water in aqueous acid solutions to afford the resulting “phenols” 45a,b:  = 2.27 × 10−4 s−1 and
= 2.27 × 10−4 s−1 and  = 6.10 × 10−4 s−1, respectively [125]. Such a reaction is negligible with mononitrobenzofurazan and mononitrobenzofuroxan analogs and almost so with 1-chloro-2,4,6-trinitrobenzene [125]. Of great significance is that the strong increase observed in SNAr reactivity goes along with an enormous increase in the stability of related σ-adducts. The hydroxy σ-adducts 47a and 47b of 4,6-dinitrobenzofurazan (DNBZ, 46a) and 4,6-dinitrobenzofuroxan (DNBF, 46b) are 1010 times thermodynamically more stable than the hydroxy adduct of TNB [127].
= 6.10 × 10−4 s−1, respectively [125]. Such a reaction is negligible with mononitrobenzofurazan and mononitrobenzofuroxan analogs and almost so with 1-chloro-2,4,6-trinitrobenzene [125]. Of great significance is that the strong increase observed in SNAr reactivity goes along with an enormous increase in the stability of related σ-adducts. The hydroxy σ-adducts 47a and 47b of 4,6-dinitrobenzofurazan (DNBZ, 46a) and 4,6-dinitrobenzofuroxan (DNBF, 46b) are 1010 times thermodynamically more stable than the hydroxy adduct of TNB [127].
From the above discussion of the water reactions, it emerges that the exceptional electrophilic character of nitrobenzofurazans and benzofuroxans corresponds to a great extension of the accessible SNAr domain. This is further demonstrated by the ease of reaction of DNBZ-Cl and DNBF-Cl with a large number of π-excessive structures of low carbon basicities such as azulene, polyalkoxybenzenes, indoles, pyrroles, and indolizines [126, 128]. All reactions proceed smoothly at room temperature, leading quantitatively to the expected substitution products, as shown in Scheme 1.2N