
Contents
About the Authors
Preface to the First Edition
Preface to the Second Edition
Acknowledgements
1 Concepts
1.1 Definition and Development of Supramolecular Chemistry
1.2 Classification of Supramolecular Host-Guest Compounds
1.3 Receptors, Coordination and the Lock and Key Analogy
1.4 Binding Constants
1.5 Cooperativity and the Chelate Effect
1.6 Preorganisation and Complementarity
1.7 Thermodynamic and Kinetic Selectivity, and Discrimination
1.8 Nature of Supramolecular Interactions
1.9 Solvation and Hydrophobic Effects
1.10 Supramolecular Concepts and Design
Summary
Study Problems
Suggested Further Reading
References
2 The Supramolecular Chemistry of Life
2.1 Biological Inspiration for Supramolecular Chemistry
2.2 Alkali Metal Cations in Biochemistry
2.3 Porphyrins and Tetrapyrrole Macrocycles
2.4 Supramolecular Features of Plant Photosynthesis
2.5 Uptake and Transport of Oxygen by Haemoglobin
2.6 Enzymes and Coenzymes
2.7 Neurotransmitters and Hormones
2.8 Semiochemistry in the Natural World
2.9 DNA
2.10 Biochemical Self-Assembly
Summary
Study Problems
References
3 Cation-Binding Hosts
3.1 Introduction to Coordination Chemistry
3.2 The Crown Ethers
3.3 The Lariat Ethers and Podands
3.4 The Cryptands
3.5 The Spherands
3.6 Nomenclature of Cation-Binding Macrocycles
3.7 Selectivity of Cation Complexation
3.8 Solution Behaviour
3.9 Synthesis: The Template Effect and High Dilution
3.10 Soft Ligands for Soft Metal Ions
3.11 Proton Binding: The Simplest Cation
3.12 Complexation of Organic Cations
3.13 Alkalides and Electrides
3.14 The Calixarenes
3.15 Carbon Donor and π-acid Ligands
3.16 The Siderophores
Summary
Study Problems
Thought Experiment
References
4 Anion Binding
4.1 Introduction
4.2 Biological Anion Receptors
4.3 Concepts in Anion Host Design
4.4 From Cation Hosts to Anion Hosts – a Simple Change in pH
4.5 Guanidinium-Based Receptors
4.6 Neutral Receptors
4.7 Inert Metal-Containing Receptors
4.8 Common Core Scaffolds
Summary
Study Problems
Thought Experiments
References
5 Ion Pair Receptors
5.1 Simultaneous Anion and Cation Binding
5.2 Labile Complexes as Anion Hosts
5.3 Receptors for Zwitterions
Summary
Study Problems
References
6 Molecular Guests in Solution
6.1 Molecular Hosts and Molecular Guests
6.2 Intrinsic Curvature: Guest Binding by Cavitands
6.3 Cyclodextrins
6.4 Molecular Clefts and Tweezers
6.5 Cyclophane Hosts
6.6 Constructing a Solution Host from Clathrate-Forming Building Blocks: The Cryptophanes
6.7 Covalent Cavities: Carcerands and Hemicarcerands
Summary
Study Problems
Thought Experiment
References
7 Solid-State Inclusion Compounds
7.1 Solid-State Host-Guest Compounds
7.2 Clathrate Hydrates
7.3 Urea and Thiourea Clathrates
7.4 Other Channel Clathrates
7.5 Hydroquinone, Phenol, Dianin’s Compound and the Hexahost Strategy
7.6 Tri-o-thymotide
7.7 Cyclotriveratrylene
7.8 Inclusion Compounds of the Calixarenes
7.9 Solid-Gas and Solid-Liquid Reactions in Molecular Crystals
Summary
Study Problems
References
8 Crystal Engineering
8.1 Concepts
8.2 Crystal Nucleation and Growth
8.3 Understanding Crystal Structures
8.4 The Cambridge Structural Database
8.5 Polymorphism
8.6 Co-crystals
8.7 Z′ > 1
8.8 Crystal Structure Prediction
8.9 Hydrogen Bond Synthons – Common and Exotic
8.10 Aromatic Rings
8.11 Halogen Bonding and Other Interactions
8.12 Crystal Engineering of Diamondoid Arrays
Summary
Study Problems
Thought Experiment
References
9 Network Solids
9.1 What Are Network Solids?
9.2 Zeolites
9.3 Layered Solids and Intercalates
9.4 In the Beginning: Hoffman Inclusion Compounds and Werner Clathrates
9.5 Coordination Polymers
Summary
Study Problem
References
10 Self-Assembly
10.1 Introduction
10.2 Proteins and Foldamers: Single Molecule Self-Assembly
10.3 Biochemical Self-Assembly
10.4 Self-Assembly in Synthetic Systems: Kinetic and Thermodynamic Considerations
10.5 Self-Assembling Coordination Compounds
10.6 Self-Assembly of Closed Complexes by Hydrogen Bonding
10.7 Catenanes and Rotaxanes
10.8 Helicates and Helical Assemblies
10.9 Molecular Knots
Summary
Study Problems
Thought Experiment
References
11 Molecular Devices
11.1 Introduction
11.2 Supramolecular Photochemistry
11.3 Information and Signals: Semiochemistry and Sensing
11.4 Molecule-Based Electronics
11.5 Molecular Analogues of Mechanical Machines
11.6 Nonlinear Optical Materials
Summary
Study Problems
References
12 Biological Mimics and Supramolecular Catalysis
12.1 Introduction
12.2 Cyclodextrins as Enzyme Mimics
12.3 Corands as ATPase Mimics
12.4 Cation-Binding Hosts as Transacylase Mimics
12.5 Metallobiosites
12.6 Haem Analogues
12.7 Vitamin B12 Models
12.8 Ion Channel Mimics
12.9 Supramolecular Catalysis
Summary
Study Problems
Thought Experiment
References
13 Interfaces and Liquid Assemblies
13.1 Order in Liquids
13.2 Surfactants and Interfacial Ordering
13.3 Liquid Crystals
13.4 Ionic Liquids
13.5 Liquid Clathrates
Summary
Study Problems
References
14 Supramolecular Polymers, Gels and Fibres
14.1 Introduction
14.2 Dendrimers
14.3 Covalent Polymers with Supramolecular Properties
14.4 Self-Assembled Supramolecular Polymers
14.5 Polycatenanes and Polyrotaxanes
14.6 Biological Self-Assembled Fibres and Layers
14.7 Supramolecular Gels
14.8 Polymeric Liquid Crystals
Summary
Study Problems
References
15 Nanochemistry
15.1 When Is Nano Really Nano?
15.2 Nanotechnology: The ‘Top Down’ and ‘Bottom Up’ Approaches
15.3 Templated and Biomimetic Morphosynthesis
15.4 Nanoscale Photonics
15.5 Microfabrication, Nanofabrication and Soft Lithography
15.6 Assembly and Manipulation on the Nanoscale
15.7 Nanoparticles
15.8 Endohedral Fullerenes, Nanotubes and Graphene
Summary
Thought Experiment
References
Index

This edition first published 2009
© 2009, John Wiley & Sons, Ltd.
Registered office
John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, United Kingdom
For details of our global editorial offices, for customer services and for information about how to apply for permission to reuse the copyright material in this book please see our website at www.wiley.com.
The right of the author to be identified as the author of this work has been asserted in accordance with the Copyright, Designs and Patents Act 1988.
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs and Patents Act 1988, without the prior permission of the publisher.
Wiley also publishes its books in a variety of electronic formats. Some content that appears in print may not be available in electronic books.
Designations used by companies to distinguish their products are often claimed as trademarks. All brand names and product names used in this book are trade names, service marks, trademarks or registered trademarks of their respective owners. The publisher is not associated with any product or vendor mentioned in this book. This publication is designed to provide accurate and authoritative information in regard to the subject matter covered. It is sold on the understanding that the publisher is not engaged in rendering professional services. If professional advice or other expert assistance is required, the services of a competent professional should be sought.
The publisher and the author make no representations or warranties with respect to the accuracy or completeness of the contents of this work and specifically disclaim all warranties, including without limitation any implied warranties of fitness for a particular purpose. This work is sold with the understanding that the publisher is not engaged in rendering professional services. The advice and strategies contained herein may not be suitable for every situation. In view of ongoing research, equipment modifications, changes in governmental regulations, and the constant flow of information relating to the use of experimental reagents, equipment, and devices, the reader is urged to review and evaluate the information provided in the package insert or instructions for each chemical, piece of equipment, reagent, or device for, among other things, any changes in the instructions or indication of usage and for added warnings and precautions. The fact that an organisation or Website is referred to in this work as a citation and/or a potential source of further information does not mean that the author or the publisher endorses the information the organisation or Website may provide or recommendations it may make. Further, readers should be aware that Internet Websites listed in this work may have changed or disappeared between when this work was written and when it is read. No warranty may be created or extended by any promotional statements for this work. Neither the publisher nor the author shall be liable for any damages arising herefrom.
Library of Congress Cataloging-in-Publication Data
Steed, Jonathan W., 1969-
Supramolecular chemistry / Jonathan W. Steed, Jerry L. Atwood. – 2nd ed.
p. cm.
Includes bibliographical references and index.
ISBN 978-0-470-51233-3 (cloth) – ISBN 978-0-470-51234-0 (pbk. :
alk. paper) 1. Supramolecular chemistry. I. Atwood, J. L. II. Title.
QD878.S74 2008
547’.1226--dc22
2008044379
A catalogue record for this book is available from the British Library.
ISBN: 978-0-470-51233-3 (Hbk)
ISBN: 978-0-470-51234-0 (Pbk)
In loving memory of Joan Edwina Steed, 1922–2008
About the Authors
Jonathan W. Steed was born in London, UK in 1969. He obtained his B.Sc. and Ph.D. degrees at University College London, working with Derek Tocher on coordination and organometallic chemistry directed towards inorganic drugs and new metal-mediated synthesis methodologies. He graduated in 1993, winning the Ramsay Medal for his Ph.D. work. Between 1993 and 1995 he was a NATO postdoctoral fellow at the University of Alabama and University of Missouri, working with Jerry Atwood. In 1995 he was appointed as a Lecturer at Kings College London and in 1998 he was awarded the Royal Society of Chemistry Meldola Medal. In 2004 he joined Durham University where he is currently Professor of Inorganic Chemistry. As well as Supramolecular Chemistry (2000) Professor Steed is co-author of the textbook Core Concepts in Supramolecular Chemistry and Nanochemistry (2007) and more than 200 research papers. He has published a large number of reviews, book chapters and popular articles as well as two major edited works, the Encyclopaedia of Supramolecular Chemistry (2004) and Organic Nanostructures (2008). He has been an Associate Editor of New Journal of Chemistry since 2001 and is the recipient of the Vice Chancellor’s Award for Excellence in Postgraduate Teaching (2006). His interests are in supramolecular sensing and molecular materials chemistry.

Jerry L. Atwood was born in Springfield MO, USA in 1942. He attended Southwest Missouri State University, where he obtained his B.S. degree in 1964. He carried out graduate research with Galen Stuckey at the University of Illinois, where he obtained his Ph.D. in 1968. He was immediately appointed as an Assistant Professor at the University of Alabama, where he rose through Associate Professor (1972) to full Professor in 1978. In 1994 he was appointed Professor and Chair at the University of Missouri – Columbia. Professor Atwood is the author of more than 600 scientific publications. His research interests revolve around a number of themes in supramolecular chemistry including gas storage and separation and the control of confined space. He has also worked on the self-assembly of non- covalent capsules, liquid clathrate chemistry, anion binding and fundamental solid state interactions, and is a world-renown crystal- lographer. He co-founded the journals Supramolecular Chemistry (1992) and Journal of Inclusion Phenomena (1983). He has edited an enormous range of seminal works in supramolecular chemistry including the five-volume series Inclusion Compounds (1984 and 1991) and the 11-volume Comprehensive Supramolecular Chemistry (1996). In 2000 he was awarded the Izatt-Christensen Prize in Supramolecular Chemistry

Preface to the First Edition
Supramolecular chemistry is one of the most popular and fastest growing areas of experimental chemistry and it seems set to remain that way for the foreseeable future. Everybody’s doing it! Part of the reason for this is that supramolecular science is aesthetically appealing, readily visualised and lends itself to the translation of everyday concepts to the molecular level. It might also be fair to say that supramolecular chemistry is a very greedy topic. It is highly interdisciplinary in nature and, as a result, attracts not just chemists but biochemists, biologists, environmental scientists, engineers, physicists, theoreticians, mathematicians and a whole host of other researchers. These supramolecular scientists are people who might be described as goal-orientated in that they cross the traditional boundaries of their discipline in order to address specific objectives. It is this breadth that gives supramolecular chemistry its wide allure, and sometimes leads to grumbling that ‘everything seems to be supramolecular these days’. This situation is aided and abetted by one of the appealing but casual definitions of supramolecular chemistry as ‘chemistry beyond the molecule’, which means that the chemist is at liberty to study pretty much any kind of interaction he or she pleases – except some covalent ones. The situation is rather reminiscent of the hubris of some inorganic chemists in jokingly defining that field as ‘the chemistry of all of the elements except for some of that of carbon’.
The funny thing about supramolecular chemistry is that despite all of this interest in doing it, there aren’t that many people who will actually teach it to you. Most of today’s practitioners in the field, including the present authors, come from backgrounds in other disciplines and are often self-taught. Indeed, some people seem as if they’re making it up as they go along! As university academics, we have both set up undergraduate and postgraduate courses in supramolecular chemistry in our respective institutions and have found that there are a lot of people wanting to learn about the area. Unfortunately there is rather little material from which to teach them, except for the highly extensive research literature with all its jargon and fashions. The original idea for this book came from a conversation between us in Missouri in the summer of 1995. Very few courses in ‘supramol,’ existed at the time, but it was clear that they would soon be increasingly common. It was equally clear that, with the exception of Fritz Vögtle’s 1991 research-level book, there was nothing by way of a teaching textbook of the subject out there. We drew up a contents list, but there the idea sat until 1997. Everybody we talked to said there was a real need for such a book; some had even been asked to write one. It finally took the persuasive powers of Andy Slade from Wiley to bring the book to fruition over the summers of 1998 and 1999. We hope that now we have written a general introductory text for supramolecular chemistry, many more courses at both undergraduate and postgraduate level will develop in the area and it will become a full member of the pantheon of chemical education. It is also delightful to note that Paul Beer, Phil Gale and David Smith have recently written a short primer on supramolecular chemistry, which we hope will be complementary to this work.
In writing this book we have been very mindful of the working title of this book, which contained the words ‘an introduction’. We have tried to mention all of the key systems and to explain in detail all of the jargon, nomenclature and concepts pertaining to the field. We have not tried to offer any kind of comprehensive literature review (for which purpose JLA has co-edited the 11 volumes of Comprehensive Supramolecular Chemistry). What errors there are will be, in the main, ones of over-simplification in an attempt to make accessible many very complicated, and often still rapidly evolving, topics. To the many fine workers whose insights we may have trivialised we offer humble apology. We hope that the overwhelming advantages will be the excitement of the reader who can learn about any or all aspects of this hydra-like field of chemistry either by a tobogganing plunge from cover to cover, or in convenient, bite-sized chunks.
Preface to the Second Edition
Since the publication of the first edition of Supramolecular Chemistry in 2000 the field has continued to grow at a tremendous pace both in depth of understanding and in the breadth of topics addressed by supramolecular chemists. These developments have been made possible by the creativity and technical skill of the international community and by continuing advances in instrumentation and in the range of techniques available. This tremendous activity has been accompanied by a number of very good books particularly at more advanced levels on various aspects of the field, including a two-volume encyclopaedia that we edited.
In this book we have tried to sample the entire field, bringing together topical research and clear explanations of fundamentals and techniques in a way that is accessible to final year undergraduates in the chemical sciences, all the way to experienced researchers. We have been very gratified by the reception afforded the first edition and it is particularly pleasing to see that the book is now available in Russian and Chinese language editions. For a short while we attempted to keep the book current by updating our system of key references on a web site; however it has become abundantly clear that a major overhaul of the book in the form of a refreshed and extended second edition is necessary. We see the strengths of the book as its broad coverage, the care we have tried to take to explain terms and concepts as they are encountered, and perhaps a little of our own personal interpretation and enthusiasm for the field that we see evolving through our own research and extensive contact with colleagues around the world. These strengths we have tried to build upon in this new edition while at the same time ameliorating some of the uneven coverage and oversimplifications of which we may have been guilty.
The original intent of this book was to serve as a concise introduction to the field of supramolecular chemistry. One of us (JWS) has since co-authored a short companion book Core Concepts in Supramolecular Chemistry and Nanochemistry that fulfils that role. We have therefore taken the opportunity to increase the depth and breadth of the coverage of this longer book to make it suitable for, and hopefully useful to, those involved at all stages in the field. Undergraduates encountering Supramolecular Chemistry for the first time will find that we have included careful explanations of core concepts building on the basics of synthetic, coordination and physical organic chemistry. At the same time we hope that senior colleagues will find the frontiers of the discipline well represented with plenty of recent literature. We have retained the system of key references based on the secondary literature that feedback indicates many people found useful, but we have also extended the scope of primary literature references for those wishing to undertake more in-depth reading around the subjects covered. In particular we have tried to take the long view both in temporal and length scales, showing how ‘chemistry beyond the molecule’ continues to evolve naturally and seamlessly into nanochemistry and molecular materials chemistry.
We have added a great deal to the book in this new edition including new chapters and subjects (e.g. supramolecular polymers, microfabrication, nanoparticles, chemical emergence, metal-organic frameworks, ion pairs, gels, ionic liquids, supramolecular catalysis, molecular electronics, polymorphism, gas sorption reactions, anion-π interactions… the list of exciting new science is formidable). We have also extensively updated stories and topics that are a part of ongoing research with new results published since 2000. The book retains some of the ‘classics’ which no less striking and informative for being a little long in the tooth these days. As before we apologise to the many fine colleagues whose work we did not include. The objective of the book is to cover the scope of the field with interesting and representative examples of key systems but we cannot be comprehensive. We feel this second edition is more complete and balanced than the first edition and we have really enjoyed putting it together. We hope you enjoy it too.
Jonathan W. Steed, Durham, UK
Jerry L. Atwood, Columbia, Missouri, USA
Acknowledgements
Our thanks go to the many fine students, researchers and colleagues who have passed through our groups over the years, whose discussions have helped to both metaphorically and literally crystallize our thinking on this rapidly evolving field. Many colleagues in both Europe and the USA have been enormously helpful in offering suggestions and providing information. In particular we are grateful to Jim Tucker, Mike Hannon, Jim Thomas and the late Fred Armitage for their help in getting the ball rolling and constructive comments on the first edition. The second edition has benefited tremendously from input by Kirsty Anderson and Len Barbour, and we are also very grateful to Len for the brilliant X-Seed which has made the crystallographic diagrams much easier to render. David Turner also provided some excellent diagrams. We thank Graeme Day for useful information on crystal structure calculation and a number of colleagues for providing artwork or additional data, particularly Sir Fraser Stoddart, John Ripmeester, Peter Tasker, Travis Holman and Bart Kahr. Beth Dufour, Rebecca Ralf and Hollie Budge, Andy Slade, Paul Deards, Richard Davies and Gemma Valler at Wiley have worked tirelessly to bring the book to the standard and accessibility it needs to have. JWS is very grateful to Durham University for providing a term of research leave which made this book so much easier to write, and we are both as ever indebted to the many fine co-workers who have passed through our labs over the years who make chemistry such an enjoyable subject to work in.
About the Front Cover
The front cover shows two views of the Lycurgus cup – a 4th century Roman chalice made of dichroic glass impregnated with nanoparticles made of gold-silver alloy. When viewed under normal lighting conditions the cup appears green but if light is shone through the glass the nanoparticles impart a gorgeous crimson colour. The chemistry of metallic nanoparticles remains a highly topical field in supramolecular chemistry. (Images courtesy of the British Museum, London, UK).
Website
Powerpoint slides of all figures from this book, along with the answers to the problems, can be found at http://www.wiley.com/go/steed
‘Mankind is divisible into two great classes: hosts and guests.’
Max Beerbohm (b. 1872), Hosts and Guests
 Lehn, J.-M., ‘Supramolecular chemistry and self-assembly special feature: Toward complex matter: Supramolecular chemistry and self-organization’, Proc. Nat. Acad. Sci. USA, 2002, 99, 4763–4768.
Lehn, J.-M., ‘Supramolecular chemistry and self-assembly special feature: Toward complex matter: Supramolecular chemistry and self-organization’, Proc. Nat. Acad. Sci. USA, 2002, 99, 4763–4768.
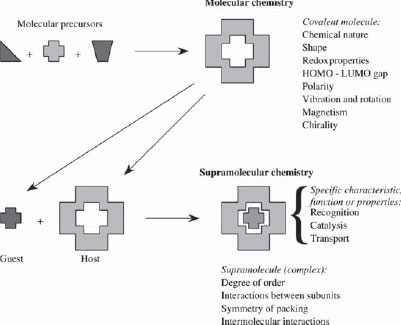
Supramolecular chemistry has been defined by one of its leading proponents, Jean-Marie Lehn, who won the Nobel Prize for his work in the area in 1987, as the ‘chemistry of molecular assemblies and of the intermolecular bond’. More colloquially this may be expressed as ‘chemistry beyond the molecule’. Other definitions include phrases such as ‘the chemistry of the non-covalent bond’ and ‘non-molecular chemistry’. Originally supramolecular chemistry was defined in terms of the non-covalent interaction between a ‘host’ and a ‘guest’ molecule as highlighted in Figure 1.1, which illustrates the relationship between molecular and supramolecular chemistry in terms of both structures and function.
These descriptions, while helpful, are by their nature noncomprehensive and there are many exceptions if such definitions are taken too literally. The problem may be linked to the definition of organometallic chemistry as ‘the chemistry of compounds with metal-to-carbon bonds’. This immediately rules out Wilkinson’s compound, RhCl(PPh3)3, for example, which is one of the most important industrial catalysts for organometallic transformations known in the field. Indeed, it is often the objectives and thought processes of the chemist undertaking the work, as much as the work itself, which determine its field. Work in modern supramolecular chemistry encompasses not just host-guest systems but also molecular devices and machines, molecular recognition, so called ‘self-processes’ such as self-assembly and self-organisation and has interfaces with the emergence of complex matter and nanochemistry (Section 1.10). The rapid expansion in supramolecular chemistry over the past 25 years has resulted in an enormous diversity of chemical systems, both designed and accidentally stumbled upon, which may lay some claim, either in concept, origin or nature, to being supramo-lecular. In particular, workers in the field of supramolecular photochemistry have chosen to adopt a rather different definition of a supramolecular compound as a group of molecular components that contribute properties that each component possesses individually to the whole assembly (covalent or non-covalent). Thus an entirely covalent molecule comprising, for example, a chromophore (light-absorbing moiety), spacer and redox centre might be thought of as supramolecular because the chromophore and redox centre are able to absorb light, or change oxidation state, whether they form part of the supermolecule or not (see Chapter 11). Similarly, much recent work has focused on the development of self-assembling synthetic pathways towards large molecules or molecular arrays. These systems often self-assemble using a variety of interactions, some of which are clearly non-covalent (e.g. hydrogen bonds) and some of which possess a significant covalent component (e.g. metal-ligand interactions, see Chapter 10). Ultimately these self-assembly reactions and the resulting self-organisation of the system rely solely on the intrinsic information contained in the structure of the molecular components and hence there is an increasing trend towards the study and manipulation of intrinsic ‘molecular information’. This shift in emphasis is nothing more than a healthy growth of the field from its roots in host-guest chemistry to encompass and inform a much broader range of concepts and activities.
Figure 1.1 Comparison between the scope of molecular and supramolecular chemistry according to Lehn.1
Kyba, E. P., Helgeson, R. C., Madan, K., Gokel, G. W., Tarnowski, T. L., Moore, S. S. and Cram, D. J., ‘Host-guest complexation. 1. Concept and illustration’, J. Am. Chem. Soc., 1977, 99, 2564–2571.
If we regard supramolecular chemistry in its simplest sense as involving some kind of (non-covalent) binding or complexation event, we must immediately define what is doing the binding. In this context we generally consider a molecule (a ‘host’) binding another molecule (a ‘guest’) to produce a ‘host-guest’ complex or supermolecule. Commonly the host is a large molecule or aggregate such as an enzyme or synthetic cyclic compound possessing a sizeable, central hole or cavity. The guest may be a monatomic cation, a simple inorganic anion, an ion pair or a more sophisticated molecule such as a hormone, pheromone or neurotransmitter. More formally, the host is defined as the molecular entity possessing convergent binding sites (e.g. Lewis basic donor atoms, hydrogen bond donors etc.). The guest possesses divergent binding sites (e.g. a spherical, Lewis acidic metal cation or hydrogen bond acceptor halide anion). In turn a binding site is defined as a region of the host or guest capable of taking part in a non-covalent interaction. The host–guest relationship has been defined by Donald Cram (another Supramolecular Chemistry Nobel Laureate)2 as follows:
Complexes are composed of two or more molecules or ions held together in unique structural relationships by electrostatic forces other than those of full covalent bonds … molecular complexes are usually held together by hydrogen bonding, by ion pairing, by π-acid to π-base interactions, by metal-to-ligand binding, by van der Waals attractive forces, by solvent reorganising, and by partially made and broken covalent bonds (transition states). High structural organisation is usually produced only through multiple binding sites… A highly structured molecular complex is composed of at least one host and one guest component… A host–guest relationship involves a complementary stereoelectronic arrangement of binding sites in host and guest… The host component is defined as an organic molecule or ion whose binding sites converge in the complex… The guest component as any molecule or ion whose binding sites diverge in the complex…
This description might well be generalised to remove the word ‘organic’, since more recent work has revealed a wealth of inorganic hosts, such as zeolites (Section 9.2) and polyoxometallates (Section 9.5.2), or mixed metal-organic coordination compounds (e.g. Section 5.2), which perform similar functions and may be thought of under the same umbrella. The host–guest binding event may be likened to catching a ball in the hand. The hand, acting as the host, envelops the ball providing a physical (steric) barrier to dropping it (disassociation). This analogy falls down at the electronic level, however, since there is no real attractive force between hand and ball. Host and guest molecules and ions usually experience an attractive force between them and hence a stabilising binding free energy. The analogy does serve to introduce the term ‘inclusion chemistry’, however (the ball is included in the hand), hence the inclusion of one molecular in another.
One key division within supramolecular host–guest chemistry in its general sense relates to the stability of a host–guest complex in solution. The field of clathrate, or more generally, inclusion, chemistry, relates to hosts that are often only stable in the solid (crystalline) state and disassociate on dissolution in a solvent. Gas hydrates, urea clathrates and a wide variety of crystalline solvates (Chapter 7) fall into this category. On the other hand, molecular hosts for ions such as the crown ethers, cryptands and spherands (Chapter 3), or hosts for neutral molecules such as the carcerands and cryptophanes (Chapter 6), display significant binding both in the solid state and in solution. We should also note that there exist purely liquid-phase phenomena, such as liquid crystals and liquid clathrates, that have no direct solid-state analogies (Chapter 13).
Supramolecular chemistry, as it is now defined, is a young discipline dating back to the late 1960s and early 1970s. However, its concepts and roots, and indeed many simple (and not-so-simple) supramolecular chemical systems, may be traced back almost to the beginnings of modern chemistry itself. An illustrative (although necessarily subjective and non-comprehensive) chronology is given in Table 1.1. Much of supramolecular chemistry has sprung from developments in macrocyclic chemistry in the mid-to-late 1960s, particularly the development of macrocyclic ligands for metal cations. Four systems of fundamental importance may be identified, prepared by the groups of Curtis, Busch, Jäger and Pedersen, three of which used the Schiff base condensation reaction of an aldehyde with an amine to give an imine (Section 3.10.6). Conceptually, these systems may be seen as a development of naturally occurring macrocycles (ionophores, hemes, porphyrins etc.). To these may be added the work of Donald Cram on macrocyclic cyclophanes (which dates back to the early 1950s) and, subsequently, on spherands and carcerands, and the tremendous contribution by Jean-Marie Lehn who prepared the cryptands in the late 1960s and has since gone on to shape many of the recent developments in the field.
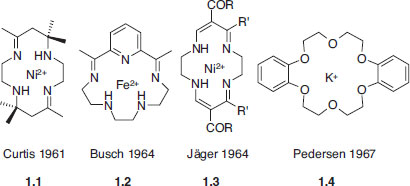
Table 1.1 Timeline of supramolecular chemistry.
| 1810 | – | Sir Humphry Davy: discovery of chlorine hydrate |
| 1823 | – | Michael Faraday: formula of chlorine hydrate |
| 1841 | – | C. Schafhäutl: study of graphite intercalates |
| 1849 | – | F. Wöhler: β-quinol H2S clathrate |
| 1891 | – | Villiers and Hebd: cyclodextrin inclusion compounds |
| 1893 | – | Alfred Werner: coordination chemistry |
| 1894 | – | Emil Fischer: lock and key concept |
| 1906 | – | Paul Ehrlich: introduction of the concept of a receptor |
| 1937 | – | K. L. Wolf: the term Übermoleküle is coined to describe organised entities arising from the association of coordinatively saturated species (e.g. the acetic acid dimer) |
| 1939 | – | Linus Pauling: hydrogen bonds are included in the groundbreaking book The Nature of the Chemical Bond |
| 1940 | – | M. F. Bengen: urea channel inclusion compounds |
| 1945 | – | H. M. Powell: X-ray crystal structures of β-quinol inclusion compounds; the term ‘clathrate’ is introduced to describe compounds where one component is enclosed within the framework of another |
| 1949 | – | Brown and Farthing: synthesis of [2.2]paracyclophane |
| 1953 | – | Watson and Crick: structure of DNA |
| 1956 | – | Dorothy Crowfoot Hodgkin: X-ray crystal structure of vitamin B12 |
| 1959 | – | Donald Cram: attempted synthesis of cyclophane charge transfer complexes with (NC)2C=C(CN)2 |
| 1961 | – | N.F. Curtis: first Schiff’s base macrocycle from acetone and ethylene diamine |
| 1964 | – | Busch and Jäger: Schiff’s base macrocycles |
| 1967 | – | Charles Pedersen: crown ethers |
| 1968 | – | Park and Simmons: Katapinand anion hosts |
| 1969 | – | Jean-Marie Lehn: synthesis of the first cryptands |
| 1969 | – | Jerry Atwood: liquid clathrates from alkyl aluminium salts |
| 1969 | – | Ron Breslow: catalysis by cyclodextrins |
| 1973 | – | Donald Cram: spherand hosts produced to test the importance of preorganisation |
| 1978 | – | Jean-Marie Lehn: introduction of the term ‘supramolecular chemistry’, defined as the ‘chemistry of molecular assemblies and of the intermolecular bond’ |
| 1979 | – | Gokel and Okahara: development of the lariat ethers as a subclass of host |
| 1981 | – | Vögtle and Weber: podand hosts and development of nomenclature |
| 1986 | – | A. P. de Silva: Fluorescent sensing of alkali metal ions by crown ether derivatives |
| 1987 | – | Award of the Nobel prize for Chemistry to Donald J. Cram, Jean-Marie Lehn and Charles J. Pedersen for their work in supramolecular chemistry |
| 1996 | – | Atwood, Davies, MacNicol & Vögtle: publication of Comprehensive Supramolecular Chemistry containing contributions from many key groups and summarising the development and state of the art |
| 1996 | – | Award of the Nobel prize for Chemistry to Kroto, Smalley and Curl for their work on the chemistry of the fullerenes |
| 2003 | – | Award of the Nobel prize for Chemistry to Peter Agre and Roderick MacKinnon for their discovery of water channels and the characterisation of cation and anion channels, respectively. |
| 2004 | – | J. Fraser Stoddart: the first discrete Borromean-linked molecule, a landmark in topological synthesis. |
As it is practised today, supramolecular chemistry is one of the most vigorous and fast-growing fields of chemical endeavour. Its interdisciplinary nature has brought about wide-ranging collaborations between physicists, theorists and computational modellers, crystallographers, inorganic and solid-state chemists, synthetic organic chemists, biochemists and biologists. Within the past decade Supramolecular chemistry has fed into very exciting new research in nanotechnology and at the interface between the two lies the area of nanochemistry (Chapter 15). The aesthetically pleasing nature of supramolecular compounds and the direct links established between the visualisation, molecular modelling and practical experimental behaviour of hosts and their complexes has fuelled increasing enthusiasm in the area to the extent that it is now a full member of the pantheon of scientific disciplines.
Vogtle, F., Supramolecular Chemistry, John Wiley & Sons, Ltd: Chichester, 1991.
One of the first formal definitions of a supramolecular cage-like host–guest structure was proposed by H. M. Powell at the University of Oxford in 1948. He coined the term ‘clathrate’, which he defined as a kind of inclusion compound ‘in which two or more components are associated without ordinary chemical union, but through complete enclosure of one set of molecules in a suitable structure formed by another’. In beginning to describe modern host–guest chemistry it is useful to divide host compounds into two major classes according to the relative topological relationship between guest and host. Cavitands may be described as hosts possessing permanent intramolecular cavities. This means that the cavity available for guest binding is an intrinsic molecular property of the host and exists both in solution and in the solid state. Conversely, clathrands are hosts with extramolecular cavities (the cavity essentially represents a gap between two or more host molecules) and is of relevance only in the crystalline or solid state. The host–guest aggregate formed by a cavitand is termed a cavitate, while clathrands form clathrates. We can also distinguish a third situation in which two molecules associate using non-covalent forces but do not fit the descriptions of ‘host’ and ‘guest’. Under these circumstances we talk about the self-assembly of a mutually complementary pair (or series) of molecules. The distinction between the two host classes and self-assembly is illustrated schematically in Figure 1.2.
A further fundamental subdivision may be made on the basis of the forces between host and guest. If the host–guest aggregate is held together by primarily electrostatic interactions (including ion-dipole, dipole-dipole, hydrogen bonding etc.) the term complex is used. On the other hand, species held together by less specific (often weaker), non-directional interactions, such as hydrophobic, van der Waals or crystal close-packing effects, are referred to by the terms cavitate and clathrate. Some examples of the use of this nomenclature are shown in Table 1.2. The distinctions between these classes are blurred and often the word ‘complex’ is used to cover all of these phenomena. Within these broad classifications a number of intermediate types exist; indeed, it is often very much a matter of opinion as to exactly what the classification of a given material might be. The nomenclature should act as a conceptual framework helping the chemist to describe and visualise the systems being handled, rather than a restrictive and rigid series of ‘phyla’.
Behr, J. P., The Lock and Key Principle. The State of the Art –100 Years on, John Wiley & Sons, Inc.: New York, 1994.
Host–guest (or receptor–substrate) chemistry is based upon three historical concepts:
Figure 1.2 Schematic illustrating the difference between a cavitate and a clathrate: (a) synthesis and conversion of a cavitand into a cavitate by inclusion of a guest into the cavity of the host molecule; (b) inclusion of guest molecules in cavities formed between the host molecules in the lattice resulting in conversion of a clathrand into a clathrate; (c) synthesis and self-assembly of a supramolecular aggregate that does not correspond to the classical host–guest description.
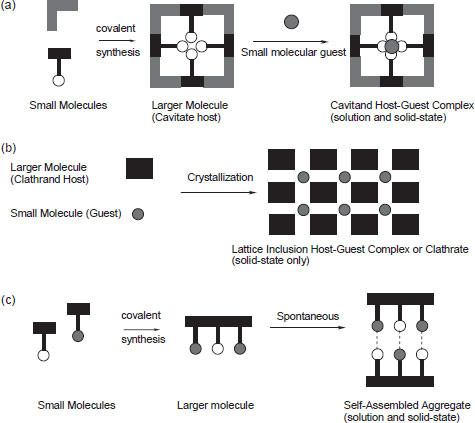
Table 1.2 Classification of common host–guest compounds of neutral hosts.
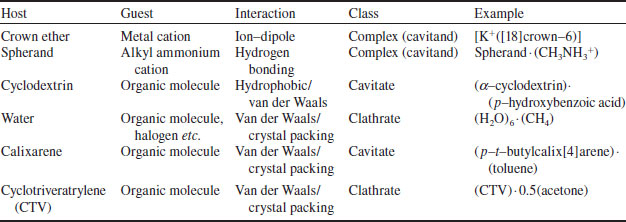
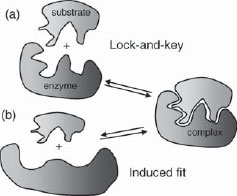
Figure 1.3 (a) Rigid lock and key and (b) induced fit models of enzyme–substrate binding.
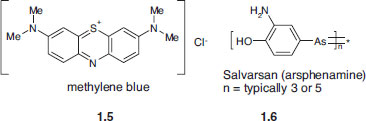
These three concepts arose essentially independently of one another and it was to be many years before the various disciplines in which they were born grew together to give birth to the highly interdisciplinary field of supramolecular chemistry. Ehrlich, for example, was working on the treatment of a range of infectious diseases. As part of his work he noticed that the dye methylene blue has a surprising affinity for some living cells, staining them an intense blue (his tutor Robert Koch had used methylene blue (1.5) to discover the tubercle bacillus, and Ehrlich had a ready supply of this synthetic dye from Farbwerke Hoechst, who had been manufacturing it since 1885). ‘If only certain cells are coloured,’ reasoned Ehrlich, ‘then may there not be dyestuffs which colour only the carriers of illnesses and at the same time destroy them without attacking the body’s own cells?’ Ehrlich eventually went on to develop the arsenic-based anti-syphilis drug Salvarsan (arsphenamine, 1.6) in 1910,3 one of the most effective drugs known for that disease. In the process he became the founder of modern chemotherapy.
The marrying of the fields of coordination chemistry, chemotherapy and enzymology was finally spurred on by the advent of modern instrumental and synthetic techniques, and not least by the dramatic developments in organic synthesis, which was born as a discipline in itself in 1828 with Friedrich Wöhler’s synthesis of urea from ammonium cyanate. In the course of the development of supramolecular chemistry, enormous progress has been made on quantifying the details of receptors with an affinity for guests which fit inside them. The lock and key image especially has suffered successive waves of modification by the concepts of cooperativity, preorganisation and complementarity, solvation and the very definition of ‘molecular shape’ as we will see in the following sections. In particular, in enzyme catalysis, the lock-and-key image has been replaced by the ‘induced fit’ theory of Daniel Koshland4 in which both enzyme and substrate (host and guest) undergo significant conformational changes upon binding to one another (Figure 1.3b). It is these conformational changes that allow the enzymatic catalytic rate acceleration since the substrate is commonly more like the reaction transition state in its bound form than in its unbound form. The occurrence of a conformational change upon guest binding is in fact a very common phenomenon both in biological chemistry, where it lies at the heart of ‘trigger’ processes such as muscle contraction and synaptic response, and in supramolecular chemistry.
Connors, K. A., Binding Constants, John Wiley & Sons, Ltd: Chichester, 1987.
The thermodynamic stability of a host–guest (e.g. metal-macrocycle) complex in a given solvent (often water or methanol) at a given temperature is gauged by measurement of the binding constant, K. Strictly the binding constant is dimensionless, but it is often calculated approximately using concentrations and thus has units of dm3 mol−1, or M−1, for a 1:1 complex. The binding constant is also known by the terms formation constant, Kf, association constant, Ka or stability constant, Ks. In biological systems the dissociation constant, Kd, is commonly used. This quantity is the reciprocal of the binding constant and has units of concentration. The Kd value is sometimes useful because it is a direct measure of the concentration below which a complex such as a drug-receptor complex will dissociate. The binding constant is the main method by which host-guest affinity in solution is assessed and so it is of fundamental importance in supramolecular chemistry and so it is worth spending some time looking into its proper definition and usage. Ignoring activity effects, the binding constant is merely the equilibrium constant for the reaction shown in Equation 1.1 (e.g. between a metal, M, and host ligand, L, in water):
(1.1) 
(1.2) 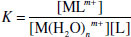
Thus a large binding constant corresponds to a high equilibrium concentration of bound metal, and hence a more stable metal–macrocycle complex. Typical binding constants for crown ethers and alkali metal cations in water are in the range 101−102. In methanol, this increases up to 106 for [K([18]crown-6)]+.* The binding constant for K+ and [2.2.2]cryptand is about 1010. Some other examples are given in Table 1.3.
Table 1.3 Binding constants for a range of complexation processes.
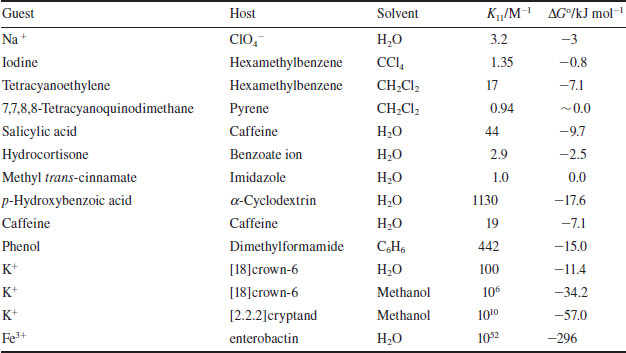
If a sequential process involving the binding of more than one metal ion is involved, then two K values may be measured for the 1:1 and 1:2 complexes, respectively: K11 and K12 (e.g. binding of two Na+ ions by dibenzo[30]crown-10).
(1.3) 
(1.4) 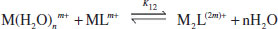
(1.5) 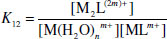
In these circumstances, an overall binding constant, β12 may be defined for the overall process, the individual K values are then known as the stepwise binding constants:
(1.6) 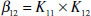
(1.7) 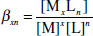
Magnitudes of binding constants can vary widely, so they are often reported as log K, hence:
(1.8) 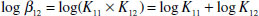
The subscript numbers in stepwise binding constant notation refer to the ratio of one complexing partner to another, thus in a multi-step process the association of the host with the first guest might be denoted K11, while the association of the resulting 1:1 complex with a further guest to produce a 1:2 complex has an equilibrium constant K12 etc. Strictly speaking it is only possible to take a logarithm of a dimensionless quantity (i.e. logs can only come from a number, not something with units) but we have to remember that the strict definition of a binding constant is based on the activities of the chemical species, not their concentrations. The activity (a) of a chemical species, i, is its effective concentration for the purposes of mass action, ai = γiCi/CΘ where Ci is the concentration of i, CΘ is equal to 1 mol dm−3 if Ci is given in mol dm−3 and γ is the activity coefficient, a factor that accounts for deviations from ideal behaviour. In approximate assessment of binding constants in supramolecular chemistry we make the approximation that γi = 1 and, activity (dimensionless) ≈ concentration.
Because binding constants are thermodynamic parameters, they are related to the free energy of the association process according to the Gibbs equation: ΔG° = −RT ln K. (R = gas constant, 8.314 J K−1 mol−1, T = temperature in Kelvin) Thus the general affinity of a host for a guest under specific conditions (solvent, temperature etc.) may be given either in terms of K or −ΔGo values. In energy terms, complexation free energies may range from magnitudes of 20 to 100 kJ mol−1 (5 to 25 kcal mol−1; 1 kJ = 4.184 kcal) for alkali metal cation complexes. A large K value of about 1010 corresponds to a −ΔGo of about 57 kJ mol−1 (13 kcal mol−1). Some very general examples of the magnitudes of binding constants and their corresponding complexation free energies are given in Table 1.3.
Binding constants may also be defined in terms of the rate constants (k) of the complexation and decomplexation reactions:
(1.9) 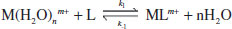
(1.10) 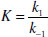
J. Polster and H. Lachmann, Spectrometric Titrations, VCH: Weinheim, 1989.
In principle, binding constants may be assessed by any experimental technique that can yield information about the concentration of a complex, [HostGuest], as a function of changing concentration of the host or guest. In practice the following methods are in common use. In every case a concentration range must be chosen such that there is an equilibrium between significant amounts of bound and free host and guest, limiting the range of binding constants that can be measured by a particular technique. If binding by the target host is too strong then a competing host is sometimes added in order to reduce the apparent (measured) binding constant according to the difference in guest affinity between the two hosts. The true affinity can then be extrapolated from a knowledge of the binding constant of the guest for the host with the lower affinity.
In the case of macrocycles that are susceptible to protonation (e.g. the cryptands with their basic tertiary amine nitrogen bridgeheads), the protonation constants (and hence pKa values) may be determined readily using pH (glass) electrodes to monitor a simple acid–base titration. Initially this will give the acid dissociation constant (pKa) of the ligand’s conjugate acid, HL+).5 Addition of a metal cation will perturb the macrocycle’s basicity (ability to bind one or more protons) by competition between the metal ion and H+ for the ligand lone pair(s) and hence will affect the shape of the titration curves.
Scheme 1.1