
Preface
Abbreviations
Chapter 1 Gene manipulation in the post-genomics era
Introduction
Gene manipulation involves the creation and cloning of recombinant DNA
The genomics era began in earnest in 1995 with the complete sequencing of a bacterial genome
Outline of the rest of the book
Part I Fundamental Techniques of Gene Manipulation
Chapter 2 Basic techniques
Introduction
Three technical problems had to be solved before in vitro gene manipulation was possible on a routine basis
A number of basic techniques are common to most gene-cloning experiments
Gel electrophoresis is used to separate different nucleic acid molecules on the basis of their size
Blotting is used to transfer nucleic acids from gels to membranes for further analysis
Southern blotting is the method used to transfer DNA from agarose gels to membranes so that the compositional properties of the DNA can be analyzed
Northern blotting is a variant of Southern blotting that is used for RNA analysis
Western blotting is used to transfer proteins from acrylamide gels to membranes
A number of techniques have been devised to speed up and simplify the blotting process
The ability to transform E. coli with DNA is an essential prerequisite for most experiments on gene manipulation
Electroporation is a means of introducing DNA into cells without making them competent for transformation
The ability to transform organisms other than E. coli with recombinant DNA enables genes to be studied in different host backgrounds
The polymerase chain reaction (PCR) has revolutionized the way that biologists manipulate and analyze DNA
The principle of the PCR is exceedingly simple
RT-PCR enables the sequences on a mRNA molecule to be amplified as DNA
The basic PCR is not efficient at amplifying long DNA fragments
The success of a PCR experiment is very dependent on the choice of experimental variables
By using special instrumentation it is possible to make the PCR quantitative
There are a number of different ways of generating fluorescence in quantitative PCR reactions
It is now possible to amplify whole genomes as well as gene segments
Useful website
Chapter 3 Cutting and joining DNA molecules
Cutting DNA molecules
Joining DNA molecules
Chapter 4 Basic biology of plasmid and phage vectors
Plasmid biology and simple plasmid vectors
Bacteriophage λ
DNA cloning with single-stranded DNA vectors
Chapter 5 Cosmids, phasmids, and other advanced vectors
Introduction
Vectors for cloning large fragments of DNA
Specialist-purpose vectors
Chapter 6 Gene-cloning strategies, 96 Introduction
Introduction
Genomic DNA libraries are generated by fragmenting the genome and cloning overlapping fragments in vectors
The PCR can be used as an alternative to genomic DNA cloning
Complementary DNA (cDNA) libraries are generated by the reverse transcription of mRNA
The PCR can be used as an alternative to cDNA cloning
Many different strategies are available for library screening
Difference cloning exploits differences in the abundance of particular DNA fragments
Chapter 7 Sequencing genes and short stretches of DNA
Chapter 8 Changing genes: site-directed mutagenesis and protein engineering
Protein engineering
Chapter 9 Bioinformatics
Introduction
Databases are required to store and cross-reference large biological datasets
Sequence analysis of genomic DNA involves the de novo identification of genes and other features
Caution must be exercised when using purely in silico methods to annotate genomes
Sequencing also provides new data for molecular phylogenetics
Part II Manipulating DNA in Microbes, Plants, and Animals
Chapter 10 Cloning in bacteria other than Escherichia coli
Cloning in Gram-negative bacteria other than E. coli
Cloning in Gram-positive bacteria
Cloning in Archaea
Chapter 11 Cloning in Saccharomyces cerevisiae and other fungi
Cloning and manipulating large fragments of DNA
Chapter 12 Gene transfer to animal cells, 218 Introduction
Introduction
There are four major strategies for gene transfer to animal cells
There are several chemical transfection techniques for animal cells but all are based on similar principles
Physical transfection techniques have diverse mechanisms
Cells can be transfected with either replicating or non-replicating DNA
Three types of selectable marker have been developed for animal cells
Plasmid vectors for the transfection of animal cells contain modules from bacterial and animal genes
DNA can be delivered to animal cells using bacterial vectors
Chapter 13 Genetic manipulation of animals
Introduction
Three major methods have been developed for the production of transgenic mice
ES cells can be used for gene targeting in mice
Applications of genetically modified mice
Applications of gene targeting
Standard transgenesis methods are more difficult to apply in other mammals and birds
Nuclear transfer technology can be used to clone animals
Gene transfer to Xenopus can result in transient expression or germline transformation
Gene transfer to fish is generally carried out by microinjection, but other methods are emerging
Gene transfer to fruit flies involves the microinjection of DNA into the pole plasma
Chapter 14 Gene transfer to plants
Introduction
Plant tissue culture is required for most transformation procedures
There are four major strategies for gene transfer to plant cells
Agrobacterium-mediated transformation
Direct DNA transfer to plants
Gene targeting in plants
In planta transformation minimizes or eliminates the tissue culture steps usually needed for the generation of transgenic plants
Plant viruses can be used as episomal expression vectors
Chapter 15 Advanced transgenic technology
Introduction
Inducible expression systems allow transgene expression to be controlled by physical stimuli or the application of small chemical modulators
Recombinant inducible systems are built from components that are not found in the host animal or plant
Site-specific recombination allows precise manipulation of the genome in organisms where gene targeting is inefficient
Many strategies for gene inactivation do not require the direct modification of the target gene
Gene inhibition is also possible at the protein level
Part III Genome Analysis, Genomics, and Beyond
Chapter 16 The organization and structure of genomes
The organization of nuclear DNA in eukaryotes
Chapter 17 Mapping and sequencing genomes
Sequencing genomes
Chapter 18 Comparative genomics
Comparative genomics of bacteria
Comparative genomics of organelles
Comparative genomics of eukaryotes
Chapter 19 Large-scale mutagenesis and interference
Introduction
Genome-wide gene targeting is the systematic approach to large-scale mutagenesis
Genome-wide random mutagenesis is a strategy applicable to all organisms
Insertional mutagenesis in invertebrates
Libraries of knock-down phenocopies can be created by RNA interference
Chapter 20 Analysis of the transcriptome
Introduction
The transcriptome is the collection of all messenger RNAs in the cell
Steady-state mRNA levels can be quantified directly by sequence sampling
DNA microarray technology allows the parallel analysis of thousands of genes on a convenient miniature device
As transcriptomics technology matures, standardization of data processing and presentation become important challenges
Expression profiling with DNA arrays has permeated almost every area of biology
Chapter 21 Proteomics I - Expression analysis and characterization of proteins
Introduction
Protein expression analysis is more challenging than mRNA profiling because proteins cannot be amplified like nucleic acids
There are two major technologies for protein separation in proteomics
Mass spectrometry is used for protein characterization
Protein microarrays can also be used for expression analysis
Chapter 22 Proteomics II - Analysis of protein structures
Introduction
Structural proteomics has required developments in structural analysis techniques and bioinformatics
International structural proteomics initiatives have been established to solve protein structures on a large scale
Chapter 23 Proteomics III - Protein interactions, 453 Introduction
Introduction
Protein interactions can be inferred by a variety of genetic approaches
New methods based on comparative genomics can also infer protein interactions
Traditional biochemical methods for protein interaction analysis cannot be applied on a large scale
Library-based screening methods allow the large-scale analysis of binary interactions
Systematic analysis of protein complexes can be achieved by affinity purification and mass spectrometry
Interaction screening produces large data sets which require extensive bioinformatic support
Chapter 24 Metabolomics and global biochemical networks
Introduction
Part IV Applications of Gene Manipulation and Genomics
Chapter 25 Applications of genomics: understanding the basis of polygenic disorders and identifying quantitative trait loci
Investigating discrete traits in outbreeding populations (genetic diseases of humans)
Investigating quantitative trait loci (QTLs) in inbred populations
Understanding responses to drugs (pharmacogenomics)
Chapter 26 Applications of recombinant DNA technology
Introduction
Theme 1: Producing useful molecules
Theme 2: Improving agronomic traits by genetic modification
Theme 3: Using genetic modification to study, prevent, and cure disease
References
Appendix: the genetic code and single-letter amino acid designations
Index

© 2006 Blackwell Publishing
BLACKWELL PUBLISHING
350 Main Street, Malden, MA 02148-5020, USA
9600 Garsington Road, Oxford OX4 2DQ, UK
550 Swanston Street, Carlton, Victoria 3053, Australia
The rights of Sandy Primrose and Richard Twyman to be identified as the Authors of this Work have been asserted in accordance with the UK Copyright, Designs, and Patents Act 1988.
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs, and Patents Act 1988, without the prior permission of the publisher.
This material was originally published in two separate volumes: Principles of Gene Manipulation, 6th edition (2001) and Principles of Genetic Analysis and Genomics, 3rd edition (2003).
First published 1980
Second edition published 1981
Third edition published 1985
Fourth edition published 1989
Fifth edition published 1994
Sixth edition published 2001
Seventh edition published 2006
1 2006
Library of Congress Cataloging-in-Publication Data
Primrose, S.B.
Principles of gene manipulation and genomics / S.B. Primrose and R.M. Twyman.—7th ed.
p.;cm.
Rev. ed. of: Principles of gene manipulation. 6th ed. 2001 and: Principles of genome analysis and genomics / Sandy B. Primrose, Richard M. Twyman. 3rd ed. 2003.
Includes bibliographical references and index.
ISBN 1-4051-3544-1 (pbk.: alk. paper) 1. Genetic engineering. 2. Genomics. 3. Gene mapping. 4. Nucleotide sequence.
[DNLM: 1. Genetic Engineering. 2. Base Sequence. 3. Chromosome Mapping. 4. DNA, Recombinant. 5. Genomics. QH 442 P952pa 2006] I. Twyman, Richard M. II. Primrose, S.B. Principles of gene manipulation. III. Primrose, S. B. Principles of genome analysis and genomics. IV. Title.
QH442.O42 2006
660.6’5—dc22
2005018202
A catalogue record for this title is available from the British Library.
For further information on
Blackwell Publishing, visit our website:
www.blackwellpublishing.com
Preface
The first edition of Principles of Gene Manipulation was published over 25 years ago when the recombinant DNA era was in its infancy and the idea of sequencing the entire human genome was inconceivable. In writing the first edition, the aim was to explain a new and rapidly growing technology. The basic philosophy was to present the principles of gene manipulation, and its associated techniques, in sufficient detail to enable the non-specialist reader to understand them. However, as the techniques became more sophisticated and advanced, so the book grew in size and complexity. Eventually, recombinant DNA technology advanced to the stage where the sequencing and analysis of entire genomes became possible. This gave rise to a whole new biological discipline, known as genomics, with its own principles and associated techniques. From this emerged the first edition of another book, Principles of Genome Analysis, whose title changed to Principles of Genome Analysis and Genomics in its third edition to reflect the rapid growth of post-sequencing technologies aiming at the large-scale analysis of gene function. It is now five years since the draft human genome sequence was published and we are reaching the stage where the technologies of gene manipulation and genomics are becoming increasingly integrated. Genome mapping and sequencing technologies borrow extensively from the early recombinant DNA technologies of library construction, cloning, and amplification using the polymerase chain reaction; gene transfer to microbes, animals, and plants is now widely used for the functional analysis of genomes; and the applications of genomics and recombinant DNA are becoming difficult to separate.
This new edition, entitled Principles of Gene Manipulation and Genomics, therefore unites the themes covered formerly by the two separate books and provides for the first time a fully integrated approach to the principles and practice of gene manipulation in the context of the genomics era. As in previous editions of the two books, we have written the text at an advanced undergraduate level, assuming a basic knowledge of molecular biology and genetics but no knowledge of recombinant DNA technology or genomics. However, we are aware that the book is favored not only by newcomers to the field but also by experts, and we have tried to remain faithful to both audiences with our coverage. As before we have not changed the level at which the book is written nor the general style, but we have divided the book into sections to enable the book to be used in different ways by different readers.
The basic methodologies are presented in the first part of the book, which is devoted to cloning in Escherichia coli, while more advanced gene-transfer techniques (applying to other microbes and to animals and plants) are presented in the second part. The reader who has read and understood the material in the first part, or already knows it, should have no difficulty in understanding any of the material in the second part of the book. The third part moves from the basic gene-manipulation technologies to genomics, transcriptomics, proteomics, and metabolomics, the major branches of the high-throughput, large-scale biology that has become synonymous with the new millennium. Finally, the fourth part of the book contains two chapters that discuss how recombinant DNA technology and genomics are being applied in the fields of medicine, agriculture, diagnostics, forensics, and biotechnology.
In writing the first part of the book, we thought carefully about the inclusion of early “historical” information. Although older readers may feel that some of this material is dated, we elected to leave much of it in place because it has an important bearing on today’s methods and an understanding of it is incorrectly assumed in many of today’s publications. We have included such information where it illustrates how modern techniques and procedures have evolved, but we have tried not to catalog outmoded or redundant methods that are no longer used. This is particularly the case in the genomics section where new technologies seem to come and go every day, and few stand the test of time or become truly indispensable. We have aimed to avoid as much jargon as possible, and to explain it clearly where it is absolutely necessary. As is common in all areas of science, the principles of gene manipulation and genomics abound with acronyms and synonyms which are often confusing particularly now molecular biology is becoming increasingly commercial in both basic research and its applications. Where appropriate, we have provided lists of definitions as boxes set aside from the text. Boxes are also used to illustrate key experiments or principles, historical information, and applications. While the text is fully referenced throughout, we have also provided a list of classic papers and reviews at the end of each chapter to ease the wary reader into the scientific literature.
This book would not have been possible without the help and advice of many colleagues. Particular thanks are due to Sue Goddard and her library staff at HPA Porton for assistance with many literature searches. Sandy Primrose would like to dedicate this book to his wife Jill and Richard Twyman would like to dedicate this book to his parents, Irene and Peter, to his children Emily and Lucy, and to Liz for her endless support and encouragement.
Abbreviations
| 2DE | two-dimensional gel electrophoresis |
| Ac | Activator |
| ADME | adsorption, distribution, metabolism and excretion |
| AFBAC | affected family-based control |
| AFLP | amplified fragment length polymorphism |
| ALL | acute lymphoblastic leukemia |
| AML | acute myeloid leukemia |
| AMV | avian myeloblastosis virus |
| APL | acute promyelocytic leukemia |
| ARS | autonomously replicating sequence |
| ATRA | all-trans-retinoic acid |
| BAC | bacterial artificial chromosome |
| BCG | Bacille Calmette–Guérin |
| bFGF | basic fibroblast growth factor |
| BIND | Biomolecular Interaction Network Database |
| BLAST | Basic Local Alignment Search Tool |
| BLOSUM | Blocks Substitution Matrix |
| BMP | bone morphogenetic protein |
| bp | base pair |
| BRET | bioluminescence resonance energy transfer |
| CAPS | cleavable amplified polymorphic sequences |
| CASP | Critical Assessment of Structural Prediction |
| CATH | Class, Architecture, Topology and Homologous superfamily (database) |
| ccc DNA | covalently closed circular DNA |
| CCD | charge couple device |
| CD | circular dichroism |
| cDNA | complementary DNA |
| CEPH | Centre d’ Etude du Polymorphisme Humain |
| cfu | commonly forming unit |
| CHEF | contour-clamped homogeneous electrical field |
| CID | chemically induced dimerization Also: collision-induced dissociation |
| cM | centimorgan |
| COG | cluster of orthologous groups |
| cR | centiRay |
| cRNA | complementary RNA |
| CSSL | chromosome segment substitution line |
| ct | chloroplast |
| DALPC | direct analysis of large protein complexes |
| DAS | distributed annotation system |
| DAS | downstream activation site |
| DBM | diazobenzyloxymethyl |
| DDBJ | DNA Databank of Japan |
| DIP | Database of Interacting Proteins |
| DMD | Duchenne muscular dystrophy |
| DNA | deoxyribonucleic acid |
| dNTP | deoxynucleoside triphosphate |
| Ds | Dissociation |
| dsDNA | double-stranded DNA |
| dsRNA | double-stranded RNA |
| EGF | epidermal growth factor |
| ELISA | enzyme-linked immunosorbent sandwich assay |
| EMBL | European Molecular Biology Laboratory |
| ENU | ethylnitrosourea |
| EOP | efficiency of plating |
| ES | embryonic stem (cells) |
| ESI | electrospray ionization |
| EST | expressed sequence tag |
| EUROFAN | European Functional Analysis Network (consortium) |
| FACS | fluorescence-activated cell sorting |
| FEN | flap endonuclease |
| FIAU | Fialuridine (1–2′-deoxy-2′-fluoro-β-d-arabinofuranosyl-5-iodouracil |
| FIGE | field-inversion gel electrophoresis |
| FISH | fluorescence in situ hybridization |
| FPC | fingerprinted contigs |
| FRET | fluorescence resonance energy transfer |
| FSSP | Fold classification based on Structure–Structure alignment of Proteins (database) |
| GASP | Genome Annotation aSsessment Project |
| G-CSF | granulocyte colony stimulating factor |
| GeneEMAC | gene external marker-based automatic congruencing |
| GGTC | German Gene Trap Consortium |
| GST | gene trap sequence tag |
| GST | glutathione-S-transferase |
| HAT | hypoxanthine, aminopterin and thymidine |
| HDL | high-density lipoprotein |
| HERV | human endogenous retrovirus |
| HGP | Human Genome Project |
| HLA | human leukocyte antigen |
| HPRT | hypoxanthine phosphoribosyltransferase |
| HTF | HpaII tiny fragment |
| htSNP | haplotype tag single nucleotide polymorphism |
| ibd | identical by descent |
| ICAT | isotope-coded affinity tag |
| IDA | interaction defective allele |
| IEF | isoelectric focusing |
| Ihh | Indian hedgehog |
| IPTG | isopropylthio-β-d-galactopyranoside |
| IST | interaction sequence tag |
| ITCHY | incremental truncation for the creation of hybrid enzymes |
| IVET | in vivo expression technology |
| kb | kilobase |
| LCR | low complexity region |
| LD | linkage disequilibrium |
| LINE | long interspersed nuclear element |
| LOD | logarithm10 of odds |
| LTR | long terminal repeat |
| m: z | mass : charge ratio |
| MAD | multiwavelength anomalous diffraction |
| MAGE | microarray and gene expression |
| MAGE-ML | microarray and gene expression mark-up language |
| MAGE-OM | microarray and gene expression object model |
| MALDI | matrix assisted laser desorption ionization |
| MAR | matrix attachment region |
| Mb | megabase |
| MCAT | mass coded abundance tag |
| MCS | multiple cloning site |
| MDA | multiple displacement amplification |
| MGED | Microarray Gene Expression Database |
| MHC | major histocompatibility complex |
| MIAME | minimum information about a microarray experiment |
| MIP | molecularly imprinted polymer |
| MIPS | Munich Information Center for Protein Sequences |
| MM | ‘mismatch’ oligonucleotide |
| MMTV | mouse mammary tumor virus |
| MPSS | massively parallel signature sequencing |
| mRNA | messenger RNA |
| MS | mass spectrometry |
| MS/MS | tandem mass spectroscopy |
| mt | mitochondrial |
| MTM | Maize Targeted Mutagenesis project |
| Mu | Mutator |
| MudPIT | multidimensional protein identification technology |
| MuLV | Moloney murine leukemia virus |
| NCBI | National Center for Biotechnology Information |
| NDB | Nucleic Acid Databank |
| NGF | nerve growth factor |
| NIGMS | National Institute of General Medical Sciences |
| NIL | near isogenic line |
| NMR | nuclear magnetic resonance |
| NOE | nuclear Overhauser effect |
| NOESY | NOE spectroscopy |
| nt | nucleotide |
| oc DNA | open circular DNA |
| OFAGE | orthogonal-field-alternation gel electrophoresis |
| OMIM | on-line Mendelian inheritance in man |
| ORF | open-reading frame |
| ORFan | orphan open-reading frame |
| P/A | presence/absence polymorphism |
| PAC | P1-derived artificial chromosome |
| PAGE | polyacrylaminde gel electrophoresis |
| PAI | pathogenicity island |
| PAM | percentage of accepted point mutations |
| PCR | polymerase chain reaction |
| PDB | Protein Databank (database) |
| Pfam | Protein families database of alignments |
| PFGE | pulsed field gel electrophoresis |
| PM | ‘perfect match’ oligonucleotide |
| poly(A)+ | polyadenylated |
| PQL | protein quantity loci |
| PRINS | primed in situ |
| PS | position shift polymorphism |
| PSI-BLAST | Position-Specific Iterated BLAST (software) |
| PTGS | post-transcriptional gene silencing |
| PVDF | polyvinylidine difluoride |
| QTL | quantitative trait loci |
| RACE | rapid amplification of cDNA ends |
| RAGE | recombinase-activated gene expression |
| RAPD | randomly amplified polymorphic DNA |
| RARE | RecA-assisted restriction endonuclease |
| RC | recombinant congenic (strains) |
| RCA | rolling circle amplification |
| RCSB | Research Collaboratory for Structural Bioinformatics |
| rDNA/RNA | ribosomal DNA/RNA |
| REMI | restriction enzyme-mediated integration |
| RFLP | restriction fragment length polymorphism |
| RIL | recombinant inbred line |
| R-M | restriction-modification |
| RNA | ribonucleic acid |
| RNAi | RNA interference |
| RNase | ribonuclease |
| RPMLC | reverse phase microcapillary liquid chromatography |
| RRS | Ras recruitment system |
| RT-PCR | reverse transcriptase polymerase chain reaction |
| RTX | repeats in toxins |
| SAGE | serial analysis of gene expression |
| SCOP | Structural Classification of Proteins (database) |
| SCOPE | structure-based combinatorial protein engineering |
| SDS | sodium dodecyl sulfate |
| SELDI | surface-enhanced laser desorption and ionization |
| SGA | synthetic genetic array |
| SGDP | Saccharomyces Gene Deletion Project |
| Shh | sonic hedgehog |
| SILAC | stable-isotope labeling with amino acids in cell culture |
| SINE | short interspersed nuclear element |
| SINS | sequenced insertion sites |
| SISDC | sequence-independent site-directed chimeragenesis |
| SNP | single nucleotide polymorphism |
| SPIN | Surface Properties of protein–protein Interfaces (database) |
| Spm | Suppressor–mutator |
| SPR | surface plasmon resonance |
| SRCD | synchrotron radiation circular dichroism |
| SRS | sequence retrieval system |
| SRS | SOS recruitment system |
| SSLP | simple sequence length polymorphism |
| SSR | simple sequence repeat |
| STC | sequence-tagged connector |
| STM | signature-tagged mutagenesis |
| STS | sequence-tagged site |
| TAC | transformation-competent artificial chromosome |
| TAFE | transversely alternating-field electrophoresis |
| TAP | tandem affinity purification |
| TAR | transformation-associated recombination |
| T-DNA | Agrobacterium transfer DNA |
| TIGR | The Institute for Genomic Research |
| TIM | triose phosphate isomerase |
| TOF | time of flight |
| tRNA | transfer RNA |
| TUSC | Trait Utility System for Corn |
| UAS | upstream activation site |
| UPA | universal protein array |
| URS | upstream repression site |
| USPS | ubiquitin-based split protein sensor |
| UTR | untranslated region |
| VDA | variant detector array |
| VIGS | virus-induced gene silencing |
| WGA | whole-genome amplification |
| Y2H | yeast two-hybrid |
| YAC | yeast artificial chromosome |
| YCp | yeast centromere plasmid |
| YEp | yeast episomal plasmid |
| YIp | yeast integrating plasmid |
| YRp | yeast replicating plasmid |
Since the beginning of the last century, scientists have been interested in genes. First, they wanted to find out what genes were made of, how they worked, and how they were transmitted from generation to generation with the seemingly mythic ability to control both heredity and variation. Genes were initially thought of in functional terms as hereditary units responsible for the appearance of particular biological characteristics, such as eye or hair color in human beings, but their physical properties were unclear. It was not until the 1940s that genes were shown to be made of DNA, and that a workable physical and functional definition of the gene – a length of DNA encoding a particular protein – was achieved (Box 1.1). Next, scientists wanted to find ways to study the structure, behavior, and activity of genes in more detail. This required the simultaneous development of novel techniques for DNA analysis and manipulation. These developments began in the early 1970s with the first experiments involving the creation and manipulation of recombinant DNA. Thus began the recombinant DNA revolution.
The definition of recombinant DNA is any artificially created DNA molecule which brings together DNA sequences that are not usually found together in nature. Gene manipulation refers to any of a variety of sophisticated techniques for the creation of recombinant DNA and, in many cases, its subsequent introduction into living cells. In the developed world there is a precise legal definition of gene manipulation as a result of government legislation to control it. In the UK, for example, gene manipulation is defined as: “… the formation of new combinations of heritable material by the insertion of nucleic acid molecules,produced by whatever means outside the cell, into any virus, bacterial plasmid or other vector system so as to allow their incorporation into a host organism in which they do not naturally occur but in which they are capable of continued propagation.” The propagation of recombinant DNA inside a particular host cell so that many copies of the same sequence are produced is known as cloning.
Cloning was a significant breakthrough in molecular biology because it became possible to obtain homogeneous preparations of any desired DNA molecule in amounts suitable for laboratory-scale experiments. A single organism, the bacterium Escherichia coli, played the dominant role in the early years of the recombinant DNA era. This bacterium had always been a popular model system for molecular geneticists and, prior to the development of recombinant DNA technology, there were already a large number of well-characterized mutants, gene regulation was understood, and many plasmids had been isolated. It is not surprising that the first cloning experiments were undertaken in E. coli and that this organism became the primary cloning host. Subsequently, cloning techniques were extended to a range of other microorganisms, such as Bacillus subtilis, Pseudomonas spp., yeasts, and filamentous fungi, and then to higher eukaryotes. Despite these advances, E. coli remains the most widely used cloning host even today because gene manipulation in this bacterium is technically easier than in any other organism. As a result, it is unusual for researchers to clone DNA directly in other organisms. Rather, DNA from the organism of choice is first manipulated in E. coli and subsequently transferred back to the original host or another organism, as appropriate. Without the ability to clone and manipulate DNA in E. coli, the application of recombinant DNA technology to other organisms would be greatly hindered.
Until the mid-1980s, all cloning was cell-based (i.e. the DNA molecule of interest had to be introduced into E. coli or another host for amplification).
In 1983, there was a further mini-revolution in molecular biology with the invention of the polymerase chain reaction (PCR). This technique allowed DNA sequences to be amplified in vitro using pure enzymes. The great sensitivity and robustness of the PCR allows DNA to be prepared rapidly from very small amounts of starting material and material of very poor quality, but it is not as accurate as cell-based cloning and only works on relatively short DNA sequences. Therefore cell-based cloning and the PCR have complementary but overlapping uses in gene manipulation.
Although the initial cloning experiments generated a great deal of excitement, it is unlikely that any of the early workers in this field could have predicted the immense impact recombinant DNA technology would have on the progress of scientific understanding and indeed on society as a whole, particularly in the fields of medicine and agriculture. Today, gene manipulation underlies a multi-billion dollar industry, employing hundreds of thousands of people worldwide and offering solutions to some of mankind’s most intractable problems. The ability to insert new combinations of genetic material into microbes, animals, and plants offers novel ways to produce valuable small molecules and proteins; provides the means to produce plants and animals that are disease-resistant, tolerant of harsh environments, and have higher yields of useful products; and provides new methods to treat and prevent human disease.
Fig. 1.1 The impact of gene manipulation on the practice of medicine.
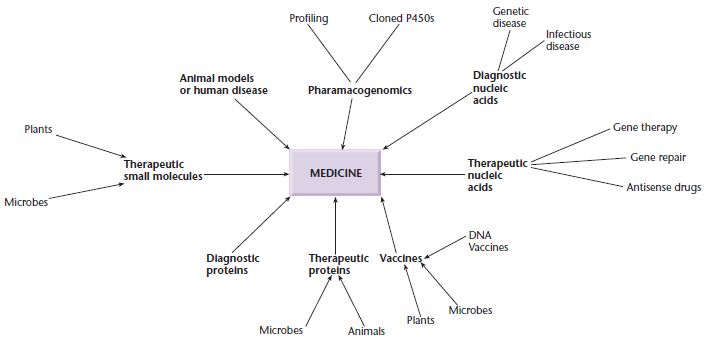
The developments in gene manipulation that have taken place in the last 30 years have revolutionized medicine by increasing our understanding of the basis of disease, providing new tools for disease diagnosis, and opening the way to the discovery or development of new drugs, treatments, and vaccines.
The first medical benefit to arise from recombinant DNA technology was the availability of significant quantities of therapeutic proteins, such as human growth hormone (HGH), which is used to treat growth defects. Originally HGH was purified from pituitary glands removed from cadavers. However, many pituitary glands are required to produce enough HGH to treat just one child. Furthermore, some children treated with pituitary-derived HGH have developed Creutzfeld–Jakob syndrome originating from cadavers. Following the cloning and expression of the HGH gene in E. coli, it became possible to produce enough HGH in a 10-liter fermenter to treat hundreds of children. Since then, many different therapeutic proteins have become available for the first time. Many of these proteins are also manufactured in E. coli but others are made in yeast or animal cells and some in plants or the milk of genetically modified animals. The only common factor is that the relevant gene has been cloned and overexpressed using the techniques of gene manipulation.
Medicine has benefited from recombinant DNA technology in other ways (Fig. 1.1). For example, novel routes to vaccines have been developed: the current hepatitis B vaccine is produced by the expression of a viral antigen on the surface of yeast cells, and a recombinant vaccine has been used to eliminate rabies from foxes in a large part of Europe. Gene manipulation can also be used to increase the levels of small molecules within microbial or plant cells. This can be done by cloning all the genes for a particular biosynthetic pathway and overexpressing them. Alternatively, it is possible to shut down particular metabolic pathways and thus redirect intermediates towards the desired end product. This approach has been used to facilitate production of chiral intermediates, antibiotics, and novel therapeutic entities. New antibiotics can also be created by mixing and matching genes from organisms producing different but related molecules in a technique known as combinatorial biosynthesis.
Gene cloning enables nucleic acid probes to be produced readily, and such probes have many uses in medicine. For example, they can be used to determine or confirm the identity of a microbial pathogen or to carry out pre- or peri-natal diagnosis of an inherited genetic disease. Increasingly, probes are being used to determine the likelihood of adverse reactions to drugs or to select the best class of drug to treat a particular illness in different groups of patients. Nucleic acids are also being used as therapeutic entities in their own right. For example, antisense nucleic acids are being used to downregulate gene expression in certain diseases, and the relatively new phenomenon of RNA interference is poised to become a breakthrough technology for the development of new therapeutic approaches. In other cases, nucleic acids are being administered to correct or repair inherited gene defects (gene therapy, gene repair) or as vaccines. In the reverse of gene repair, animals are being generated that have mutations identical to those found in human disease. These are being used as models to learn more about disease pathology and to test novel therapies.
As well as techniques for DNA cloning and transfer to new host cells, the recombinant DNA revolution spawned new technologies for gene mapping (ordering genes on chromosomes) and DNA sequencing (determining the order of bases, identified by the letters A, C, G, and T, along the DNA molecule). Within the gene itself, the order of bases determines the protein encoded by the gene by specifying the order of amino acids. Thus, DNA sequencing made it possible to work out the amino acid sequence of the encoded protein without the direct analysis of the protein itself. This was extremely useful because, at the time DNA sequencing was first developed, only the most abundant proteins in the cell could be purified in sufficient quantities to facilitate direct analysis. Further elements surrounding the coding region of the gene were identified as control regions, specifying each gene’s expression profile. As more sequence data accumulated, it became possible to identify common features in related genes, both in the coding region and the regulatory regions. This type of sequence analysis was greatly facilitated by the foundation of sequence databases, and the development of computer-aided techniques for sequence analysis and comparison, a field now known as bioinformatics. Today, DNA molecules can be scanned quickly for a whole series of structural features, e.g. restriction enzyme recognition sites, matches or overlaps with other sequences, start and stop signals for transcription and translation, and sequence repeats, using programs available on the Internet.
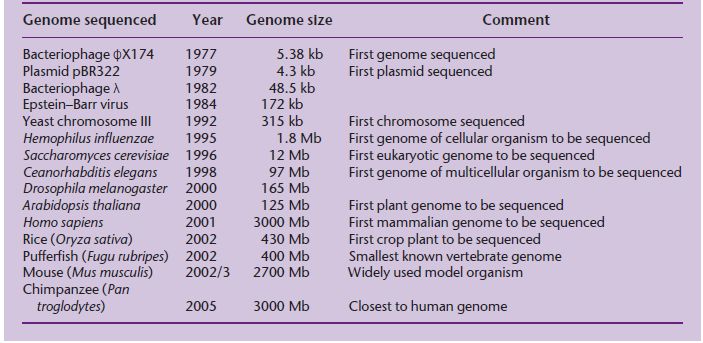
The original goal of sequencing was to determine the precise order of nucleotides in a gene, but soon the goal became the sequence of a small genome. A genome is the complete content of genetic information in an organism, i.e. all the genes and other sequences it contains. The first target was the genome of a small virus called ϕX174, then larger plasmid and viral genomes, then chromosomes and microbial genomes until ultimately the complete genomes of higher eukaryotes were sequenced (Table 1.1). In the mid-1980s, scientists began to discuss seriously how the entire human genome might be sequenced. To put these discussions in context, the largest stretch of DNA that can be sequenced in a single pass (even today) is 600–800 nucleotides and the largest genome that had been sequenced in 1985 was that of the 172-kb Epstein–Barr virus (Baer et al. 1984). By comparison, the human genome is 3000 Mb in size, over 17,000 times bigger! One school of thought was that a completely new sequencing methodology would be required, and a number of different technologies were explored but with little success. Early on, however, it was realized that existing sequencing technology could be used if a large genome could be broken down into more manageable pieces for sequencing in a highly parallel fashion, and then the pieces could be joined together again. A strategy was agreed upon in which a map of the human genome would be used as a scaffold to assemble the sequence.
Table 1.1 Timeline of genome sequencing, showing the increasing genome sizes that have been achieved.
The problem here was that in 1985 there were not enough markers, or points of reference, on the human genome map to produce a physical scaffold on which to assemble the complete sequence. Genetic maps are based on recombination frequencies, and in model organisms they are constructed by carrying out large-scale crosses between different mutant strains. The principle of a genetic map is that the further apart two loci are on a chromosome, the more likely that a crossover will occur between them during meiosis. Recombination events resulting from crossovers can be scored in genetically amenable organisms such as the fruit fly Drosophila melanogaster and yeast by looking for new combinations of the mutant phenotypes in the offspring of the cross. This approach cannot be used in human populations because it would involve setting up large-scale matings between people with different inherited diseases. Instead, human genetic maps rely on the analysis of DNA sequence polymorphisms, i.e. naturally occurring DNA sequence differences in the population which do not have an overt, debilitating effect. A major breakthrough was the development of methods for using DNA probes to identify polymorphic sequences (Botstein et al. 1980).
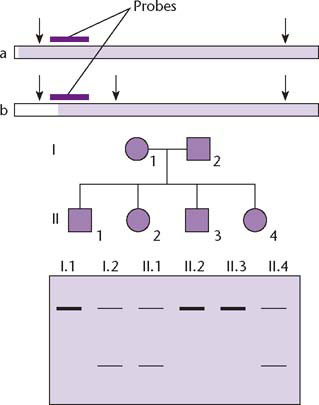
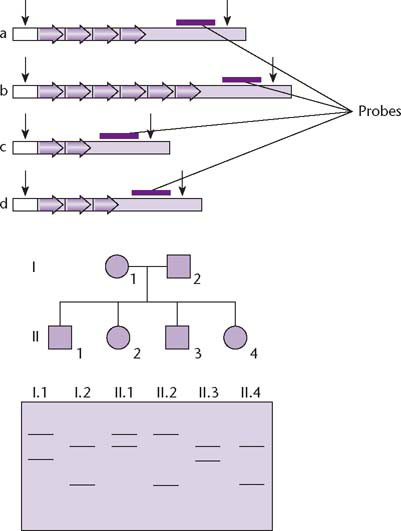
Prior to the Human Genome Project (HGP), low-resolution genetic maps had been constructed using restriction fragment length polymorphisms (RFLPs). These are naturally occurring variations that create or destroy sites for restriction enzymes and therefore generate different sized bands on Southern blots (Fig. 1.2). The Southern blot is a technique for separating DNA fragments by size, see Fig. 2.6, p. 23. The problem with RFLPs was that they were too few and too widely spaced to be of much use for constructing a framework for physical mapping – the first RFLP map had just over 400 markers and a resolution of 10 cM, equivalent to one marker for every 10 Mb of DNA (Donis-Keller et al. 1987). The necessary breakthrough came with the discovery of new polymorphic markers, known as microsatellites, which were abundant and widely dispersed in the genome (Fig. 1.3). By 1992, a genetic map based on microsatellites had been constructed with a resolution of 1 cM (equivalent to one marker for every 1 Mb of DNA) which was a suitable template for physical mapping.
Fig. 1.2 Restriction fragment length polymorphisms (RFLPs) are sequence variants that create or destroy a restriction site in DNA therefore altering the length of the restriction fragment that is detected. The top panel shows two alternative alleles, in which the restriction fragment detected by a specific probe differs in length due to the presence or absence of the middle of three restriction sites (represented by vertical arrows). Alleles a and b therefore produce hybridizing bands of different sizes in Southern blots (lower panel). This allows the alleles to be traced through a family pedigree. For example child II.2 has inherited two copies of allele a, one from each parent, while child II.4 has inherited one copy of allele a and one copy of allele b.
Unlike genetic maps, physical maps are based on real units of DNA and therefore provide a basis for sequence assembly. The physical mapping phase of the HGP involved the creation of genomic DNA libraries and the identification and assembly of overlapping clones to form contigs (unbroken series of clones representing contiguous segments of the genome). When the HGP was initiated, the highest-capacity vectors available for cloning were cosmids, with a maximum insert size of 40 kb. Because hundreds of thousands of cosmid clones would have to be screened to assemble a physical map, the HGP would not have progressed very quickly without the development of novel high-capacity vectors and methods to find overlaps between them so that clone contigs could be assembled on the genomic scaffold.
Fig. 1.3 Microsatellites are sequence variants that cause restriction fragments or PCR products to differ in length due to the number of copies of a short tandem repeat sequence, 1–12 nt in length. The top panel shows four alternative alleles, in which the restriction fragment detected by a specific probe differs in length due to a variable number of tandem repeats. All four alleles produce bands of different sizes on Southern blots (lower panel) or different sized PCR products (not shown). Unlike RFLPs, multiple allelism is common for microsatellites so the precise inheritance pattern in a family pedigree can be tracked. For example, the mother and father in the pedigree have alleles b/d and a/c, respectively (the smaller DNA fragments move further during electrophoresis). The first child, II.1, has inherited allele b from his mother and allele a from his father.
The late 1980s and early 1990s saw much debate about the desirability of sequencing the human genome. This debate often strayed from rational scientific debate into the realms of politics, personalities, and egos. Among the genuine issues raised were questions such as:
Behind the debate was a fear that sequencing the human genome was an end in itself, much like a mountaineer who climbs a new peak just because it is there.
The publicly funded Human Genome Project was officially launched in 1990, and the scientific community began to develop new strategies to enable the large-scale mapping and sequencing that were required to complete the project, strategies which centered around high-throughput, highly parallel automated sequencing. One of the benefits of this new technology development was the completion of several pilot genome projects, beginning with that of the bacterium Hemophilus influenzae (Fleischmann et al. 1995). The net effect was that by the time the human genome had been sequenced (International Human Genome Sequencing Consortium 2001, Venter et al. 2001), the complete sequence was already known for over 30 bacterial genomes plus that of a yeast (Saccharomyces cerevisiae), the fruit fly, a nematode (Caenorhabditis elegans), and a plant (Arabidopsis thaliana).
Parallel developments in the field of bioinformatics were required to handle and analyze the exponentially increasing amounts of sequence data arising from the genome projects, but bioinformatics also facilitated the development of new sequencing strategies. For example, when a European consortium set itself the goal of sequencing the entire genome of the budding yeast S. cerevisiae (15 Mb), they segmented the task by allocating the sequencing of each chromosome to different groups. That is, they subdivided the genome into more manageable parts. At the time this project was initiated there was no other way of achieving the objective and when the resulting genomic sequence was published (Goffeau et al. 1996), it was the result of a unique multi-institution collaboration. While the S. cerevisiae sequencing project was underway, a new genomic sequencing strategy was unveiled: shotgun sequencing. In this approach, large numbers of genomic fragments are sequenced and sophisticated bioinformatics algorithms used to construct the finished sequence. In contrast to the consortium approach used with S. cerevisiae, a single laboratory set up as a sequencing factory undertook shotgun sequencing.
The first success with shotgun sequencing was the complete sequence of the bacterium H. influenzae (Fleischmann et al. 1995) and this was quickly followed with the sequences of Mycoplasma genitalium (Fraser et al. 1995), Mycoplasma pneumoniae (Himmelreich et al. 1996) and Methanococcus jannaschii (Bult et al. 1996). It should be noted that H. influenzae was selected for sequencing because so little was known about it: there was no genetic map and not much biochemical data either. By contrast, S. cerevisiae was a well-mapped and well-characterized organism. As will be seen in Chapter 17, the relative merits of shotgun sequencing vs. ordered, map-based sequencing are still being debated today. Nevertheless, the fact that a major sequencing laboratory can turn out the entire sequence of a bacterium in 1–2 months shows the power of shotgun sequencing.
Fears that sequencing the human genome would be an end in itself have proved groundless. Because so many different genomes have been sequenced it is now possible to undertake comparative analyses of genomes, a topic known as comparative genomics. By comparing genomes from distantly related species we can begin to decipher the major stages in evolution. By comparing more closely related species we can begin to uncover more recent events such as genome rearrangement which have facilitated speciation (see e.g. Murphy et al.