
Table of Contents
Related Titles
Title Page
Copyright
Preface
List of Contributors
Chapter 1: Tetronic Acids
1.1 Introduction
1.2 Natural Occurrence, Biological Activities, and Biosynthesis
1.3 5-Ylidene Tetronic Natural Products
1.4 5-Monosubstituted Tetronic Natural Products
1.5 5-Disubstituted Tetronic Natural Products
1.6 5-Unsubstituted Tetronic Natural Products
1.7 Conclusions
References
Chapter 2: Recent Advances in the Field of Naturally Occurring 5,6-Dihydropyran-2-ones
2.1 Introduction
2.2 Synthetic Methodologies for 5,6-Dihydropyran-2-ones
2.3 Formation of Stereogenic Centers inside the Dihydropyrone Ring
2.4 Pharmacological Properties of Pyrones
2.5 Biosynthetic Formation of Pyrones
2.6 Syntheses of Natural 5,6-Dihydropyran-2-ones Reported during the Period from 2006 to the First Half of 2012
References
Chapter 3: β-Lactams
3.1 Introduction
3.2 Monocyclic β-Lactams
3.3 Penams
3.4 Cephalosporins
3.5 Clavulanic Acid
3.6 Carbapenems
3.7 Spiro-Fused β-Lactams
3.8 Summary
References
Chapter 4: α-Alkylidene-γ- and δ-Lactones and Lactams
4.1 Introduction
4.2 Occurrence, Biosynthesis, and Biological Activities of α-Alkylidene γ- and δ-Lactones and Lactams
4.3 Recent Advances in the Synthesis of α-Alkylidene-γ- and δ-Lactones and Lactams
4.4 Conclusions
References
Chapter 5: Medium-Sized Lactones
5.1 Introduction
5.2 Total Synthesis of Eight-Membered Lactones
5.3 Total Synthesis of Nine-Membered Lactones
5.4 Conclusions
References
Chapter 6: Macrolactones
6.1 Introduction
6.2 General Methods for the Synthesis of Macrolactones
6.3 Synthesis of Macrolides
6.4 Synthesis of Macrodiolides
6.5 Synthesis of Macrotriolides
6.6 Conclusions and Perspectives
Abbreviations
References
Chapter 7: Resorcylic Acid Lactones
7.1 Introduction – A Historical Perspective
7.2 Biosynthesis
7.3 Chemical Synthesis
7.4 Conclusion and Outlook
References
Chapter 8: Cyclic Peptides
8.1 Introduction
8.2 Synthesis of Natural Lactones and Lactams
8.3 Conclusion
References
Index
Related Titles
Dinges, J., Lamberth, C. (eds.)
Bioactive Heterocyclic Compound Classes
Pharmaceuticals
2012
ISBN: 978-3-527-33395-0
(Also available in digital formats)
Lamberth, C., Dinges, J. (eds.)
Bioactive Heterocyclic Compound Classes
Agrochemicals
2012
ISBN: 978-3-527-33396-7
(Also available in digital formats)
Hanessian, S., Giroux, S., Merner, B.L.
Design and Strategy in Organic Synthesis
From the Chiron Approach to Catalysis
2013
ISBN: 978-3-527-33391-2
Eicher, T., Hauptmann, S., Speicher, A.
The Chemistry of Heterocycles
Structure, Reactions, Synthesis, and Applications
Third Edition
2012
ISBN: 978-3-527-32868-0
Pozharskii, A. F., Soldatenkov, A., Katritzky, A. R.
Heterocycles in Life and Society
An Introduction to Heterocyclic Chemistry,
Biochemistry and Applications
Second Edition
2011
ISBN: 978-0-470-71410-2
(Also available in digital formats)
Anderson, R.R., Groundwater, P.P., Todd,~A.A., Worsley, A.A.
Antibacterial Agents \endash\, Chemistry,
Mode of Action, Mechanisms of
Resistance and Clinical Applications
2012
ISBN: 978-0-470-97244-1
(Also available in digital formats)
Diana, P., Cirrincione, G.
Biosynthesis of Heterocycles
2013
ISBN: 978-1-118-02867-4
Yudin, A.K. (ed.)
Catalyzed Carbon-Heteroatom Bond Formation
2011
ISBN: 978-3-527-32428-6
(Also available in digital formats)
Majumdar, K.C., Chattopadhyay, S.K. (eds.)
Heterocycles in Natural Product Synthesis
2011
ISBN: 978-3-527-32706-5
(Also available in digital formats)

The Editor
Prof. Tomasz Janecki
Lodz University of Technology
Department of Chemistry
Institute of Organic Chemistry
Żeromskiego 116
90-924 łód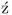
Poland
All books published by Wiley-VCH are carefully produced. Nevertheless, authors, editors, and publisher do not warrant the information contained in these books, including this book, to be free of errors. Readers are advised to keep in mind that statements, data, illustrations, procedural details or other items may inadvertently be inaccurate.
Library of Congress Card No.: applied for
British Library Cataloguing-in-Publication Data
A catalogue record for this book is available from the British Library.
Bibliographic information published by the Deutsche Nationalbibliothek
The Deutsche Nationalbibliothek lists this publication in the Deutsche Nationalbibliografie; detailed bibliographic data are available on the Internet at <http://dnb.d-nb.de>.
© 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Boschstr. 12, 69469 Weinheim, Germany.
All rights reserved. (including those of translation into other languages). No part of this book may be reproduced in any form — by photoprinting, microfilm, or any other means — nor transmitted or translated into a machine language without written permission from the publishers. Registered names, trademarks, etc. used in this book, even when not specifically marked as such, are not to be considered unprotected by law.
Print ISBN: 978-3-527-33414-8
ePDF ISBN: 978-3-527-66694-2
ePub ISBN: 978-3-527-66693-5
Mobi ISBN: 978-3-527-66692-8
oBook ISBN: 978-3-527-66691-1
Lactones and lactams are one of the best recognized classes of natural products. They can be found in many natural sources and have very diversified structure with the ring size varying from 4-membered up to 60-membered. The synthesis and chemistry of natural lactones and lactams have been extensively studied in many laboratories for a long time. The main reason for the enormous interest in these two classes of compounds arises from the fact that lactone or lactam moiety is present in a vast number of natural and synthetic compounds displaying a wide spectrum of desired biological properties. Moreover, this moiety is usually crucial for their activity. The most important application of the lactones and lactams is in the pharmaceutical industry. Currently, plenty of drugs containing these structural motifs, such as β-lactams, macrolactones, dihydropyran-2-ones or tetronic acids are used in medicinal treatment of inflammation, cancer, malaria and many other diseases. Many others are on different steps of medicinal trials. Well known are also the odorant properties of many lactones which are used in fine and functional perfumery. Musk odorants which are macrolactones or relatively simple γ- and δ-lactones like whisky, wine or Aerangis lactones are the best known examples. The second important reason for the great attention given by chemists to natural lactones and lactams is their attractiveness as chiral building blocks which are frequently used in organic synthesis. Taking all these facts into consideration, it is really surprising that, to the best of my knowledge, any comprehensive compilation dedicated to this group of natural products has not been published so far. I believe that this book fills this gap, at least to some extent.
The lactone or lactam structural motif can be found in so many natural products that it was not possible to include all of them in this book. Consequently, the selection which had to be made is, to a great extent, the arbitral decision of the editor proceeded by discussions with college chemists. The content of this book is therefore arguable and one can imagine somewhat different choice of subjects. One of the first things I had to do when starting the edition of this book was to get in touch with and secure the cooperation of the undisputable experts in specific and sometime narrow groups of lactones or lactams. Although it was a challenge in a few cases, the list of contributors to this book shows clearly that this undertaking has been successively accomplished.
The book is organized in eight chapters devoted to different classes of natural lactones and lactams and I believe that all main groups are included. The only really big class which is not discussed in this book are coumarins but fortunately many reviews devoted to the occurrence, biological activity and synthesis of this class of compounds have recently been published (see A. Yu. Fedorov, A. V. Nyuchev and I. P. Beletskaya Chemistry of Heterocyclic Compounds 2012, 48, 166, and references cited therein). All contributors to this book were asked to include in their chapters a general overview as well as information about the occurrence and biological activity of the specific class of lactones and/or lactams. However, the main part of each contribution is dedicated to the general and most recent synthetic methods leading to each class of compounds. The authors were encouraged to adhere to this general scheme, however their own ideas on the content of the chapter and personal style were fully honored.
I would like to thank all the authors and coauthors of the individual chapters for their excellent and timely contributions. Also, I would like to give special acknowledgments to the Wiley-VCH editorial staff, in particular to Anne Brennfuehrer who inspired me to take up the challenge of developing this book and guided me through all the steps of the editorial process and to Lesley Belfit who took care of all the technical aspects of book preparation.
Łód, May 2013Tomasz Janecki
List of Contributors
 eromskiego 116eromskiego 116
eromskiego 116eromskiego 116Tetronic acids belong to the class of 4-hydroxybutenolides that are characterized by a 4-hydroxy-2(5H)-furanone ring, as it can be seen in the generic structure of Scheme 1.1 [1]. This type of five-membered vinylogous acids can be met in two main tautomeric forms (1 and 2) with enol structure 1 being the one that predominates. Tetronic acid is a substructural element of many natural products of various classes such as alkaloids, terpenes, macrolides, and tannins. Two well-known natural products of this class are ascorbic acid (3) and penicillic acid (4). In many cases, its presence in natural products is connected with a wide range of significant biological properties and, therefore, the synthesis of these compounds has attracted synthetic interest for more than a century. In the following section, a synopsis of the natural occurrence, biological activities, and biosynthetic considerations of tetronic acid natural products will be attempted and a more thorough analysis of the synthetic strategies toward such compounds will be presented. Categorization of tetronic structures is based on 5-position's substitution and discussion on synthetic routes toward these compounds will be limited on total syntheses with main focus to the construction of tetronic ring.
Scheme 1.1 Generic structure of a tetronic acid and structures of ascorbic acid (3) and penicillic acid (4).
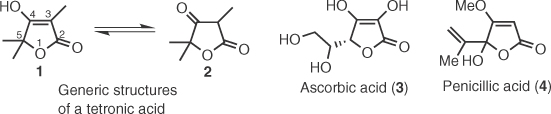
A large group of tetronic natural products isolated from numerous fungi, molds, and lichens are pulvinic acids (16) and their decarboxylated analogs, pulvinones (14) [2]. The first pulvinic derivative was isolated by Berbet in 1831 from the lichen Cetraria vulpina, whereas the first pulvinone was isolated by Gill in 1973 by the mushroom Boletus elegans [3]. Pulvinic dimers such as norbadione A (from bolete Xerocomus badius [4]) and sclerocitrin (from puffball Scleroderma citrinum [5]) have also been isolated and they have been biosynthetically linked with the dimerization of xerocomic acid by an ingenious mechanism proposed by Steglich et al. [5]. Vulpinic acid and analogs have displayed anti-inflammatory, antipyretic, and analgesic properties, but several of these compounds are cytotoxic [6]. Norbadione A, di-O-methyl atromentic acid, and derivatives exhibit marked antioxidant and radioprotective properties [7], whereas variegatic and xerocomic acids are inhibitors of cytochrome P450 enzymes [8]. In addition, norbadione A acts as a strong Cs2+ chelator and has been studied for applications in 137Cs decontamination [9]. Biosynthetically, pulvinic acids and pulvinones are related to terphenylquinones (9 and 10) and grevillins (11), two isomeric classes of natural products that stem from the dimerization of arylpyruvates 7 that directly connects them with the shikimate pathway (Scheme 1.2) [2a]. This was supported by feeding experiments that established l-phenylalanine and l-tyrosine as essential building blocks for their biosynthesis [10] as well as the identification of the atromentin's gene cluster that delineated the biogenetic relation of tyrosine with 10 [11].
Scheme 1.2 Biosynthesis of pulvinic acids (16) and pulvinones (14).
3-Acyl tetronic acids are probably the biologically most interesting class of tetronic acid natural products. For example, tetronasin [12], an ionophore antibiotic produced from Streptomyces longisporoflavus, is used as an animal-feed supplement and interferes with the selective permeation of ions across lipid bilayers causing depolarization of the membrane and death [13]. This molecule tends to bind metal ions by using its 3-acyl-tetronic moiety. RK-682, a simple 3-palmitoyl-5-hydroxymethylene tetronic acid isolated from various Actinomycetes and Streptomycetes sources [14], is a potent inhibitor of HIV-1 protease [14a] and various protein tyrosine kinases and phosphatases [14b] (including VHR and cdc25B) by acting as a phosphate mimic. In 2010, Sun et al. [15] reported the full reconstruction of RK-682 biosynthesis by using recombinant enzymes via a pathway that shares many common features with that of spirotetronate antibiotics that will be discussed later. Indications from feeding/NMR (nuclear magnetic resonance) experiments have shown that the biosynthesis of relevant agglomerins [16] may follow a similar pathway that is based on the incorporation of a glyceryl unit into the tetronic ring [17]. On the contrary, in 1962, an alternative route has been proposed by Bentley et al. [18] for the biosynthesis of 3-acyl tetronic acids isolated from molds (carlic, carolic, and carlosic acids) that involves enzymatic acylation of γ-methyl tetronic derivatives derived from C4-dicarboxylic acids.
A group of tetronic acid natural products with challenging structural patterns from both synthetic and biosynthetic viewpoints and interesting antibiotic and antitumor properties are the spirotetronic natural products. Tetrocarcin A (TCA) is produced by Micromonospora chalcea NRRL 11289 and possesses a polycyclic aglycon (tetronolide) that features a trans-decalin system and a tetronate moiety spiro linked with a cyclohexene ring and two sugar side chains heavily responsible for their biological activity [19]. TCA-induced apoptosis in tumor cells [20] can be mediated by (i) antagonizing the mitochondrial functions of proteins of the Bcl-2 family (natural apoptosis inhibitors) in HeLa cells [20a], (ii) activating the caspase-dependent cell death pathway via endoplasmic reticulum stress in lymphomas [20b,c], and (iii) inhibiting the phosphorylation of factors involved in phosphatidylinositol 3-kinase/Akt signaling in human breast cancer cells [20d]. On bioassay using experimental mouse models, TCA exhibited remarkable antitumor activities without significant myelosuppression and associated nephrotoxicity [21]. Arisostatins A and B, two tetrocarcins of similar biological properties, have been isolated in 2000 from Micromonospora sp. TP-A0316 [22]. Recent results have indicated that arisostatin A induces apoptosis in HN-4 cells by mediating loss of mitochondrial transmembrane potential, release of cytochrome C into cytosol and generation of reactive oxygen species [23]. More than 60 structurally related compounds have been reported from strains of various Gram-positive bacteria (Actinomycetes) and nearly all of them possess antibacterial and antitumor activities. Well-known examples include the first member to be discovered in this family, chlorothricin [24], as well as kijanimicin [25], lobophorins A–F [26], MM46115 [27], and versipelostatin [28]. Chlorothricin acts as an inhibitor of pyruvate carboxylase that catalyzes the anaplerotic CO2 fixation on synthetic media and it is able to inhibit cholesterol biosynthesis via the mevalomalonate pathway [29]. Kijanimicin has a broad spectrum of antimicrobial activity against Gram-positive bacteria, anaerobes, and the malaria parasite Plasmodium falciparum and also shows antitumor activity [25, 30]. Interestingly, lobophorins A and B exhibit anti-inflammatory and not antibiotic properties, based on experiments performed in the PMA-induced mouse ear edema model [26a]. Versipelostatin down-regulates the expression of GRP78, a molecular chaperone in the endoplasmic reticulum that plays an important role as a surviving factor in solid tumors because of its acquisition of a resistant mechanism against both chemotherapy and hypoglycemic stress [28, 31]. Later, it was demonstrated that versipelostatin can specifically inhibit the activation of the unfolded protein response (UPR), in response to glucose deprivation [31]. The result of versipelostatin action is enhanced sensitivity to glucose deprivation with enhanced cell death after exposure to the drug in the glucose-deprived state.
Another group of structurally related spirotetronate macrolides lacking the decalin core include spirotetronic tetramers quartromicins [32], spirohexenolides [33], and abyssomicins [34]. Spirohexenolide A displays strong cytotoxicity in the NCI-60 cell line and low toxicity in mice [33], whereas quartromicins exhibit antiviral activity against herpes simplex virus type 1, influenza virus type A, and HIV [32]. Natural atropoisomers abyssomicin C and atrop-abyssomicin C display significant antibiotic activity against a variety of Gram-positive bacteria, including methicillin- and vancomycin-resistant Staphylococcus aureus strains, with the second being the most active [34a, 35]. These molecules are the first natural products able to inhibit the p-aminobenzoic acid (pABA), a constituent of the folate pathway, by covalently binding to aminodesoxychorismate synthase via a Michael addition of a cysteine nucleophile [36].
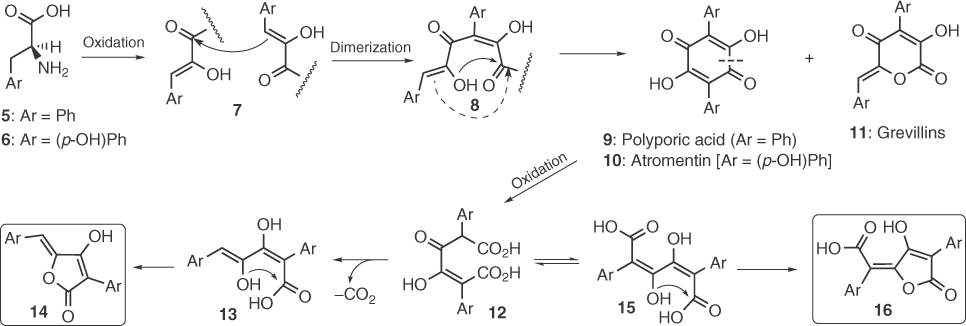
Initial results from feeding experiments complemented by the identification of the gene clusters [37] of chlorothricin [37a], kijanimicin [37b], TCA [37c], abyssomicin [37d], and quartromicin [37e] as well as the related non-spiro tetronates tetronomycin [38] and RK-682 [15] have contributed to the elucidation of a reasonable biosynthetic route for the tetronate moiety (Scheme 1.3). In particular, a polyketide unit (19), synthesized by a module type I polyketide synthase (PKS), and a glycerol derived 3-carbon unit bound to an acyl carrier protein, ACP (glyceryl-S-ACP, 18) are combined to form the tetronate moiety. The latter is synthesized by an FkbH-like glyceryl-S-ACP synthase that catalyzes d-1,3-bisphosphoglycerate dephosphorylation and transfer to ACP. In the case of RK-682, the formation of the C–C and C–O bonds of the tetronic ring seems to be mediated by the action of the same synthase, but it is not clear in what order and whether this is also the case for other tetronic natural products. In the case of kijanimicin and TCA, an FAD-dependent oxidoreductase has been suggested to be involved in the dehydration and subsequent intramolecular (or intermolecular, in the case of quartromicins) Diels–Alder reaction, but it is still not clear whether such processes are catalyzed or spontaneous (e.g., a similar gene was not found in the case of abyssomicin). It should be noted that similar Diels–Alder reactions have been proposed for the biosynthesis of spirotetronic sesterterpenes ircinianin and wistarin [39]. In this case, the unprecedented occurrence of both wistarin enantiomers in nature may imply that this process is enzyme catalyzed. Finally, feeding/NMR experiments with versipelostatin imply the intermediacy of a glycerol unit in its biosynthesis, although no genetic data are still available [40].
Scheme 1.3 Biosynthetic route toward spirotetronates.
The representative examples of this family, as shown in Figure 1.1, are pulvinic acids (X = OH), vulpinic acids (X = OMe), and pulvinones (analogs lacking carboxyl or ester group). These compounds constitute a large group of 5-arylidenetetronates and can be extracted from lichenous and higher fungal sources [2a]. Since the pioneering synthesis of vulpinic acid (24) by Volhard in 1894 [41], the total synthesis of many members of this family has been described (Figure 1.1).
Figure 1.1 Structures of naturally occurring pulvinic acids.
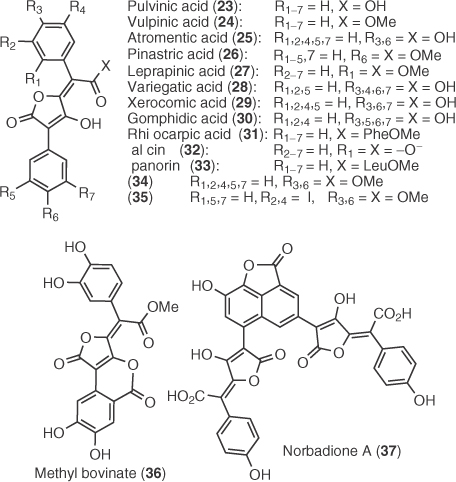
From 1894 (Volhard's syntheses of 23 and 24) until the early 1970s, hydrolysis (for pulvinic acids) or methanolysis (for vulpinic acids) of dilactones of type 38 was the main synthetic strategy (Scheme 1.4) [42]. Volhard's dilactone approach was based on the condensation of diethyl oxalate and phenyl acetonitrile and subsequent acidic hydrolysis and dehydration [41]. Asano and Kameda [43] and Akermak [44] extended the methodology for unsymmetrical dilactones by the successive use of different aryl acetonitriles in the condensation step, but the final formation of two pulvinic isomers (39 and 40, Ar1 ≠ Ar2) after dilactone opening was unavoidable. Alternative routes to access dilactones 38 have also been developed [45], with the most important being the biosynthetically inspired oxidative rearrangement of aryl hydroxyquinones such as polyporic acid 9 [45b,c] or atromentin 10 [45a]. Among the various conditions used for this oxidation, Moore's protocol [dimethyl sulfoxide/acetic anhydride (DMSO/Ac2O)] was and still is the most widely used (i.e., Steglich's synthesis of methyl bovinate (36) in 2008 [46]). Dilactone strategy has been the basis for the total synthesis of a variety of pulvinic/vulpinic acids, such as atromentic (25) [42a], pinastric (26) [42b,c], leprapinic (27) [42d], variegatic (28) [42e], xerocomic (29) [42f], gomphidic (30) [42g], rhizocarpic (31) [42h] acids, calycin (32) [44], and epanorin (33) [42h]. Nevertheless, with the only exception of Moore's synthesis of vulpinic acid based on the thermal rearrangement of azidoquinone 41 to pulvinonitrile 42 followed by hydrolysis (Scheme 1.4) [47], no regiospecific methods have been reported during 80 years after Volhard's introduction of dilactone strategy.
Scheme 1.4 Pulvinic acid synthesis from dilactones 38 and azidoquinone 41.

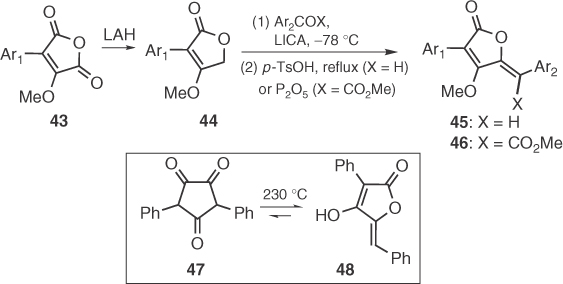
In 1975, Pattenden et al. reported the first regiospecific synthesis of permethylated pulvinic acids 46 [48] and pulvinones 45 [49] by condensation of 44 with aroyl formates and aryl aldehydes, respectively, followed by dehydration (Scheme 1.5). Interestingly, Pattenden's pioneering work allowed the first total synthesis of naturally occurring pulvinones, first isolated from natural sources in 1973 [3b] but already produced by Claisen in 1895 [50] from the thermal rearrangement of a symmetrical trione (47 → 48, Scheme 1.5).
Scheme 1.5 Syntheses of pulvinic derivatives by Pattenden and Claisen.
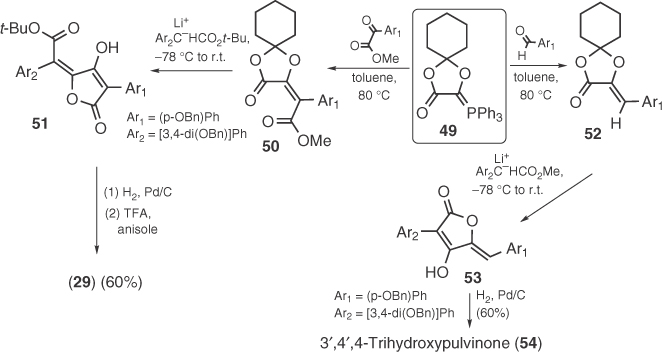
In 1984, Ramage et al. [51] have demonstrated that dioxolane phosphorane 49 can lead to pulvinic acids and pulvinones after olefination with α-ketoesters or aldehydes, respectively, and Claisen condensation with arylacetic esters (Scheme 1.6). In the case of pulvinic acids [51b], regiospecificity can be controlled by proper choice of ester groups, as this was outlined in the synthesis of xerocomic acid (29). Ramage's protocol was also used for the synthesis of multicolanic acid [51c], a tetronic metabolite found in Penicillium multicolor, which was previously synthesized by Pattenden via maleic anhydride chemistry [52]. In 2007, Bruckner's group modified Ramage's protocol by using similar phosphonates in order to replace the olefination step by a Horner–Wadsworth–Emmons (HWE) reaction [53].
Scheme 1.6 Syntheses of 29 and 54 by Ramage.
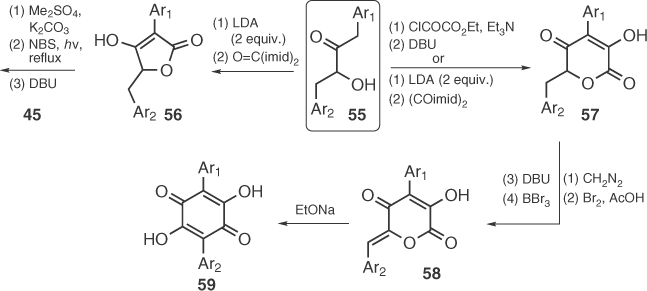
Working independently, Gill and Pattenden employed benzylacyloins (55) as synthetic precursors to obtain grevillins (58, pulvinic acids biosynthetic progenitors) and pulvinones 45, (Scheme 1.7). Synthesis of pulvinones by Gill [54] was based on a previous report by Smith's group [55] and relies on the condensation of unsymmetrical bis-benzyl acyloins with carbonyldiimidazole. Similar condensation of 55 with oxalyldiimidazole (Gill's method [54]) or esterification with ethyloxalyl chloride followed by Dieckmann condensation (Pattenden's method [56]) led to grevillins (58) that rearrange to quinones 59 [57], affording pulvinic acids after application of Moore's protocol [45c].
Scheme 1.7 Use of benzylacyloins in the synthesis of pulvinic derivatives.
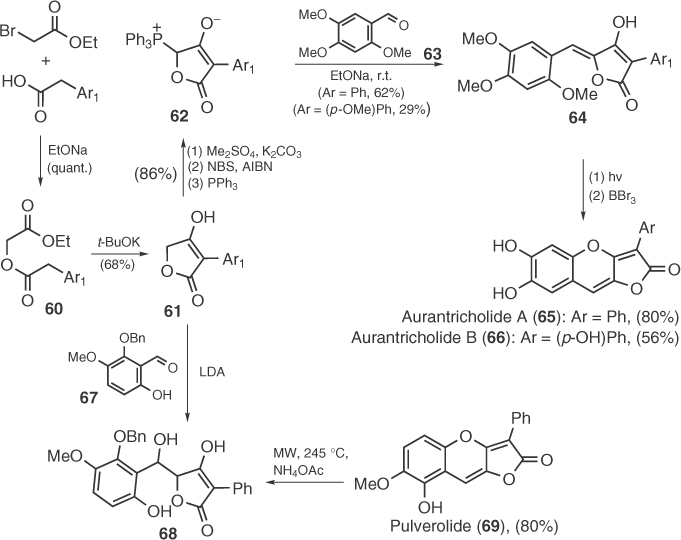
In 1985, Campbell et al. [58] employed a Dieckmann condensation strategy to the synthesis of pulvinones. By this route, Campbell prepared tetronic acid 61 that can be either transformed to phosphorane 62 and utilized in Wittig reactions or condensed with arylaldehydes that can afford pulvinones after dehydration. The first route led Steglich's group in 2000 to the total synthesis of aurantricholides A (65) and B (66), two minor pigments of toadstool Tricholoma aurantium [59], whereas the second one was used for the total synthesis of pulverolide (69), a pigment isolated from Pulveroboletus ravenelii, by Yang et al. [60] in 2010 (Scheme 1.8).
Scheme 1.8 Total syntheses of aurantricholides A and B and pulverolide.
Furthermore, the Dieckmann condensation strategy has also been employed in the synthesis of pulvinic acids [61]. Interestingly, 30 years after the first relevant report by Weinstock et al. [61a], the group of Le Gall [61b] presented a versatile route for that purpose. Le Gall improved the synthesis of tetronic acid 61, previously prepared by Campbell et al. [58], by devising a tandem transesterification/Dieckmann condensation and offered the first total synthesis of 35 after application of Pattenden's arylidenation (Scheme 1.9) [48a]. Moreover, Le Gall et al. [61c] demonstrated that the use of aryl acetates of dimethyl tartrate can lead to tetronic acid 71 that can be coupled by aryl groups after iodination and Suzuki coupling reaction, thus affording pinastric acid (26). Recent reports on improved pulvinone synthesis based on Dieckmann condensation have also appeared [62] with that of Brückner's group succeeding on the total synthesis of several naturally occurring aspulvinones (i.e., aspulvinone B, Scheme 1.9) [62a]. Brückner introduced aryl groups of aspulvinones before the Dieckmann condensation by using a Heck reaction to 2-acetoxyacrylate 72 and subsequent esterification with arylacetic acids.
Scheme 1.9 Total syntheses of aspulvinone B and pulvinic acids 26 and 35.
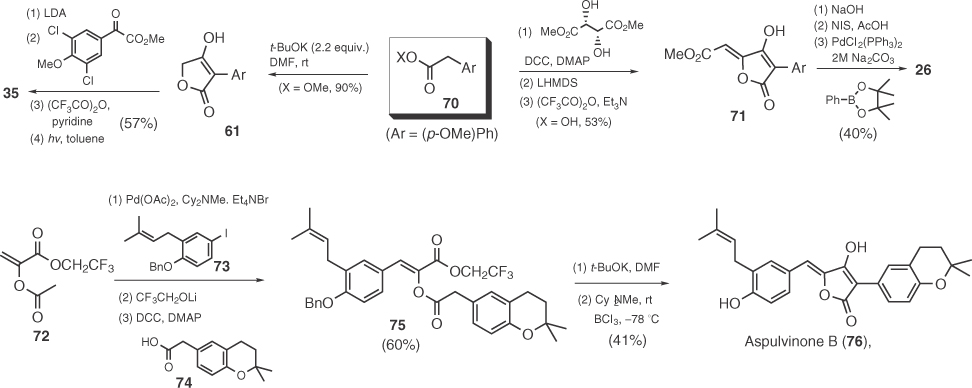
In 1991, Pattenden exploited the inherent regioselectivity of nucleophilic additions on β-methoxy maleic anhydrides to prepare gomphidic acid (30) by two different routes (Scheme 1.10) [63]. The first one involves an HWE reaction between phosphonate 80 and aryl pyruvate 81 [63b], and the second one is based on a Reformatsky-type reaction of 77 with zinc enolates derived from aryl acetate 79 [63c].
Scheme 1.10 Total syntheses of gomphidic acid by Pattenden.
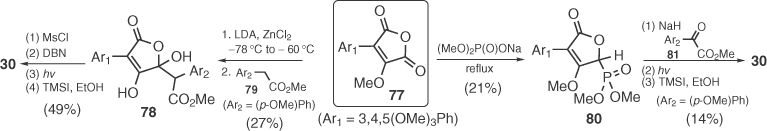
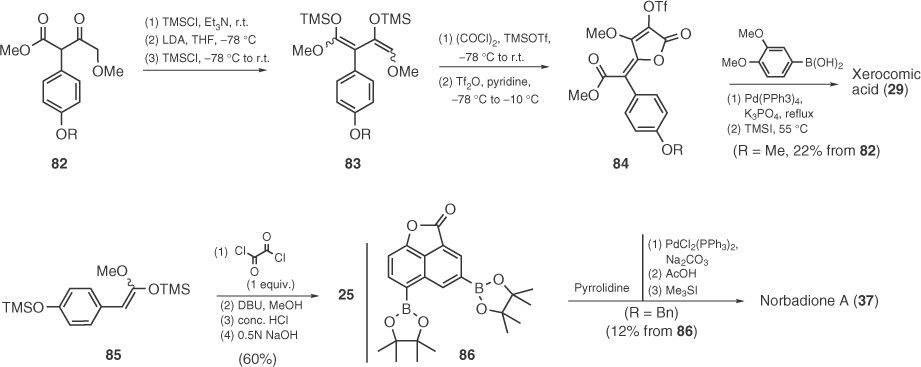
In a series of reports, Langer's group studied the synthesis of pulvinic acids via a TMSOTf-catalyzed [3+2] cyclization of 1,3-bis-(trimethylsilyloxy)-1,3-dienes (e.g., 83) with oxalyl chloride followed by Suzuki coupling of product triflates (Scheme 1.11) [64]. Langer's versatile method was applied to the synthesis of almost all natural pulvinic acids (e.g., xerocomic acid 29), including norbadione A (37) by Le Gall et al. [65]. In a modification of Langer's protocol, Mioskowski dimerized simple silyl ketene acetals (e.g., 85) to obtain symmetrical pulvinic acids via an uncatalyzed reaction with (COCl)2 (Scheme 1.11) [66].
Scheme 1.11 Application of Langer's chemistry to the synthesis of pulvinic derivatives.
In 2006, Le Gall et al. [67] presented a conceptually different approach for the synthesis of pulvinic acids, starting from commercial tetronic acid 87 (Scheme 1.12). The protocol involves application of Pattenden's arylidenylation by aroyl formates, iodination, and aryl coupling by Suzuki reaction. The utility of Le Gall's method was exemplified by the synthesis of vulpinic (24) and pinastric (26) acid.
Scheme 1.12 Le Gall's synthesis of pulvinic acids.
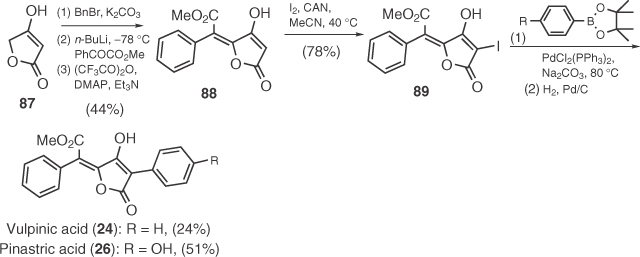
Agglomerins constitute a group of 3-acyl-5-methylidene tetronic acids with antibiotic activity [16]. Synthetic efforts toward agglomerins started by Ley and coworkers [68] who successfully applied a Pd-catalyzed acylation on stannyl tetronate 91 toward the synthesis of agglomerin A (Scheme 1.13) (92). In 2005, Schobert's group prepared agglomerins A–C (92–94) following a triphenylphosphoranylidene/glycerate 95 Wittig cyclization and a DCC-mediated 3-acylation of 96 [69], according to Yoshii's protocol (Scheme 1.13) [70].
Scheme 1.13 Total syntheses of agglomerins A–C.
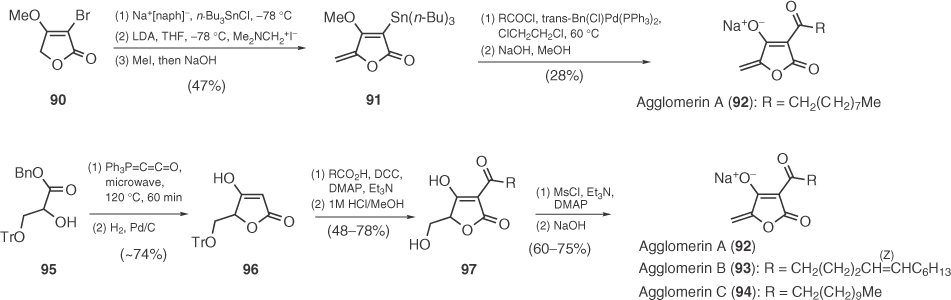
Tetronomycin (102), a structurally challenging 3-acyltetronic acid ionophore antibiotic, was isolated from a Streptomyces strain in the early 1980s [71]. The only total synthesis reported so far was accomplished in 1992 by Yoshii and coworkers (Scheme 1.14) [72]. By using as chiral building blocks the ethoxyethyl ether 98 and l-rhamnal diacetate 99, Yoshii reached aldehyde 100, a suitable precursor for the installation of the tetronic moiety. This was achieved by a reaction with the lithium anion of methyl tetronate 101 at −100 °C that led to the target after pyridinium chlorochromate (PCC) oxidation and careful deprotection.
Scheme 1.14 Total syntheses of tetronomycin.

Stemofoline alkaloids constitute a group of natural products isolated from Stemona plants that have been used in Chinese and Japanese folk medicines as cough-relief agents and insecticides [73]. These molecules are characterized by a rigid polycyclic core bearing a pendant methyl tetronate. Synthetic attempts toward these targets were mainly focused on the construction of the bridged alkaloid nucleus [74], whereas racemic total synthesis of isostemofoline (106) by Kende et al. [75] and asparagamine A (116) and isodidehydrostemofoline (117) by Overman et al. [76] have been reported. Kende's synthesis is based on an Rh-catalyzed [4+3] cycloaddition of vinyl diazoester 104 with pyrrole intermediate 103 (Scheme 1.15), whereas Overman's approach utilizes a Diels–Alder reaction between ethyl (E)-3-nitroacrylate and 108 and an aza-Cope–Mannich reaction as key steps to form the main polycyclic framework (Scheme 1.16).
Scheme 1.15 Kende's total syntheses of isostemofoline.
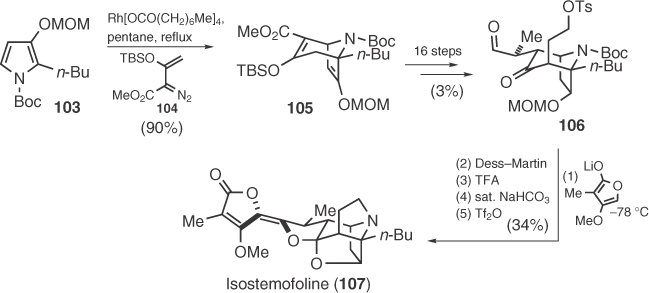
Scheme 1.16 Racemic and enantioselective syntheses of 116 and 117.
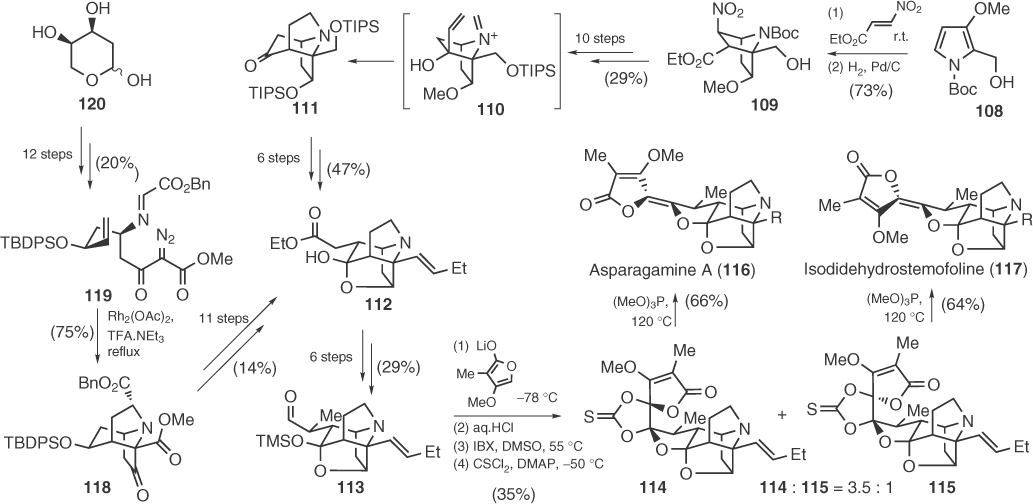
Interestingly, both researches employed the same technique to install the methyl tetronate ring, that is, addition of the lithium anion of 4-methoxy-3-methyl-2(5H)-furanone to an aldehyde (106 or 113), but Kende used a Tf2O-mediated dehydration to finish the synthesis, whereas Overman obtained the final structures after a Corey–Winter reaction to form the dialkoxy alkene unit (Scheme 1.16). In 2012, Martin et al. [77] managed to prepare enantioselectively Overman's intermediate 113 by an elegant cascade of reactions that culminates in the intramolecular dipolar cycloaddition of 119, prepared from 2-deoxy-d-ribose (120) (Scheme 1.16).
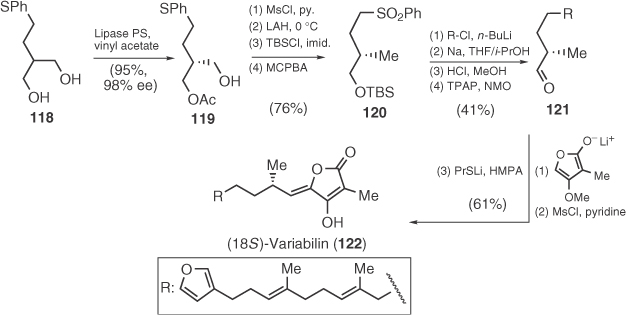
The bioactive marine furanosesterterpene tetronic acid (18S)-variabilin (122) [78] has been synthesized by Takabe et al., 30 years after its first isolation (Scheme 1.17) [79]. Stereochemical control was achieved by enzymatic desymmetrization of propanediol 118, whereas the tetronic core was introduced via an aldol condensation/mesylation/elimination sequence.
Scheme 1.17 Total synthesis of variabilin.
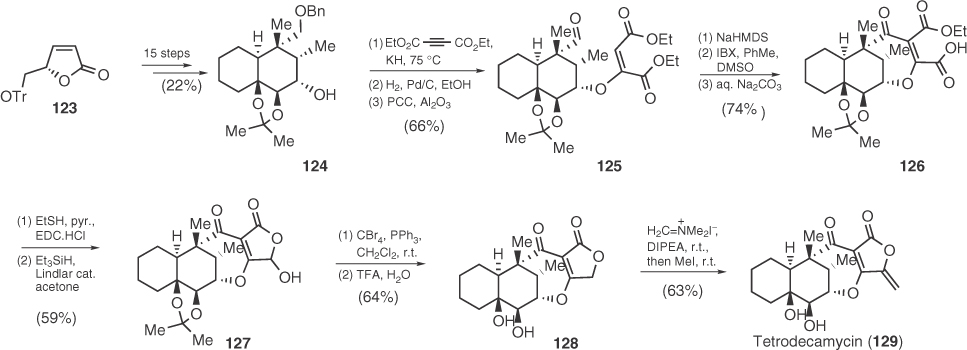
Tetrodecamycin (129) is a polyketide antibiotic isolated in 1994 from the culture broth of Streptomyces nashvillensis MJ885-mF8 and was found to exhibit promising antibacterial activity against Gram-positive bacteria [80]. Owing to its challenging decalin core structure doubly anchored with a 5-methylene tetronate, a number of model studies have been published by the groups of Paintner, Barriault, and Tchabanenko mainly focused on the decalin core of the molecule [81], whereas Paintner investigated also the attachment of tetronic ring by using a novel 3-hydroxyalkylation reaction of boron 4-methoxy-2-furanolates [82]. The first enantioselective total synthesis of tetrodecamycin reported by Tatsuta et al. [83] in 2006, included a very interesting strategy to introduce a tetronate ring on the decalin structure 124 obtained from carbohydrate derivative 123. According to Scheme 1.18, compound 124 was transformed in three steps to aldehyde 125 that was annulated to afford the third ring of the target via a sodium hexamethyldisilazide (NaHMDS)-mediated Baylis–Hillman type reaction. Finally, the inventive formation of the tetronic ring of tetrodecamycin relies on the reduction of carboxylic acid 126 that leads to hemiacetal 127 and a novel deoxygenation operation using CBr4 and PPh3.
Scheme 1.18 Total synthesis of tetrodecamycin.
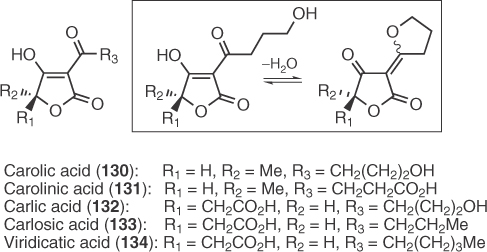
A number of fungal 5-substituted 3-acyl tetronic acid metabolites, as shown in Scheme 1.19, have attracted significant attention during the development of new synthetic protocols in the field of tetronic acids.
Scheme 1.19 Structures of carolic, carolinic, carlic, carlosic and viridicatic acids.
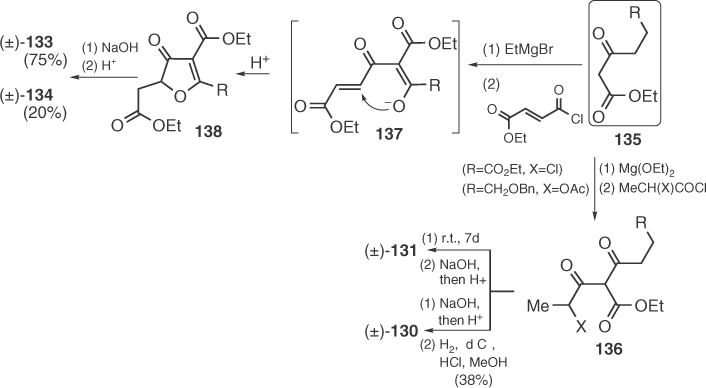
The first syntheses of racemic carolinic acid (131) by Haynes et al. [84], carolic acid (130) by Sudo et al. [85], and carlosic (133) and viridicatic (134) acids by Svendsen and Boll [86] were based on a C5–O1 disconnection of the tetronic ring. The main difference of these strategies is that cyclization by Haynes and Sudo proceeds via an intramolecular substitution, whereas Svendsen and Boll perform a Michael addition for the same purpose (Scheme 1.20). Notable feature of the latter syntheses is the alkaline-induced rearrangement of the initial condensation products (138) to the final tetronic acid metabolites 133 (R = n-propyl) and 134 (R = n-pentyl).
Scheme 1.20 First racemic syntheses of 130, 131, 133, and 134.
Aiming to a C3–C4 stereoselective disconnection approach, Bloomer and Kappler [87] managed to cyclize malic derivative 139, as shown in Scheme 1.21. Deacetylation toward 140 followed by an O-acylation/Fries rearrangement sequence with butyryl chloride/TiCl4/PhNO2 afforded (S)-carlosic acid [87b]. Following similar protocols, the syntheses of (R)-carolic acid and (±)-carlic acid have also been achieved [87a,c]. Some years later, Ley et al. [88] performed the synthesis of (S)-carlosic and (S)-carlic acids by a fluoride-induced cyclization of malic derivatives 142 and 143 derived from the transesterification of β-ketothiesters 141 with (S)-dimethyl malate (Scheme 1.21).
Scheme 1.21 Use of malic derivatives in the synthesis of 132 and 133.
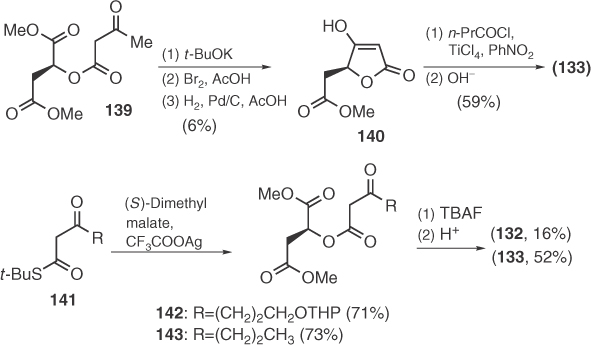
An extension of the use of malic derivatives by Mitsos et al. [89] includes the regioselective ring opening of malic anhydrides 144 by β-ketoesters 145 that successfully leads to (S)-carlosic and (S)-viridicatic acids (Scheme 1.22). Finally, Schobert and Jagusch [90] employed their methodology of constructing tetronic structures from α-hydroxyesters and Ph3P=C=C=O to the efficient synthesis of (S)-carlosic acid, as shown in Scheme 1.22.
Scheme 1.22 Synthesis of 133 and 134 by Mitsos and Schobert.
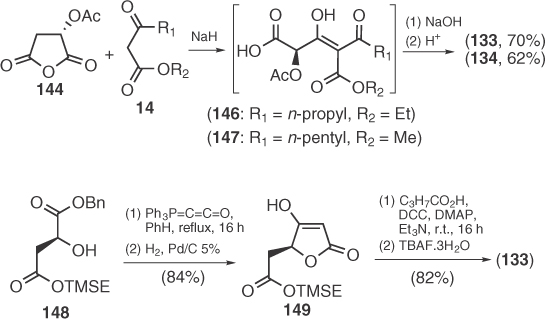
RK-682 (152), a simple 3-alkanoyl-5-hydroxymethyltetronic acid, was structurally elucidated by Sodeoka et al. [91] through its total synthesis starting from the d-mannitol derived glycerate 150 and subsequent application of Ley's synthetic protocol (Scheme 1.23) [88]. Two years later, Ohta and coworkers [92] prepared RK-682 and other related HIV-1 protease inhibitors by a modified Masamune acylation of lactone 157 (157 → 159) followed by a phenylselenylation strategy to create the double bond of the tetronate. The convergence of Ohta's strategy was recently recognized by Ramachary's group who developed an organocatalytic method for the synthesis of intermediate 156, also providing a formal synthesis of RK-682 as well as agglomerins (92–94) [93]. As RK-682 can be considered as a hydrated agglomerin-related precursor, Schobert and Jagusch [69] applied their protocol for agglomerin synthesis to the preparation of RK-682 on solid phase (Scheme 1.23).
Scheme 1.23 Various synthetic routes toward RK-682.
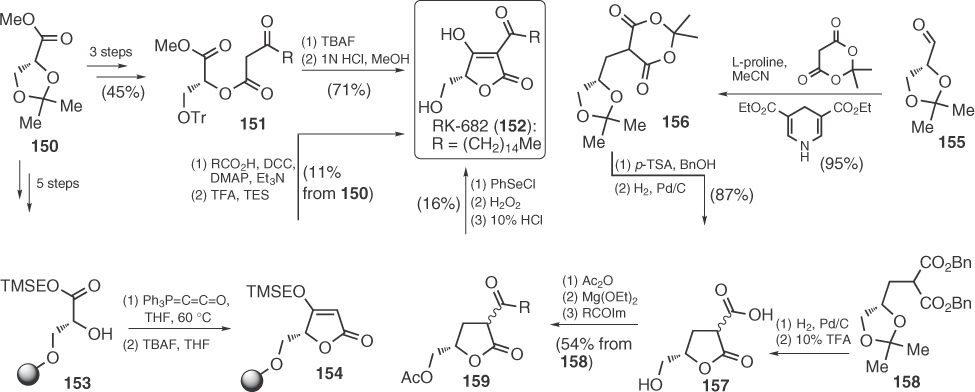
Massarilactone B (163), a bicyclic tetronate with antibacterial activity [94], was synthesized by Snider and Gao, as shown in Scheme 1.24 [95]. Key features of the synthesis are the highly diastereoselective iodoetherification of intermediate 160 and the establishment of the tetronic core at 162 through an elimination process.
Scheme 1.24 Total synthesis of massarilactone B.
Annularins F (167), G (172), and H (170) are three polyketide metabolites, recently isolated from the organic extracts of the freshwater fungus Annulatascus triseptatus [96]. The first racemic synthesis reported for the antibacterial annularin F by Hsung et al. [97] involved a condensation of 164 and 165 toward pyrone 166 that was converted to 167 in two steps (Scheme 1.25). In 2007, Reissig et al. [98] produced racemic annularin H by a synthetic route based on the addition of lithium methoxyallene to aldehyde 168 followed by Au(I)-induced cyclization (Scheme 1.25).
Scheme 1.25 Syntheses of racemic annularins F and H.
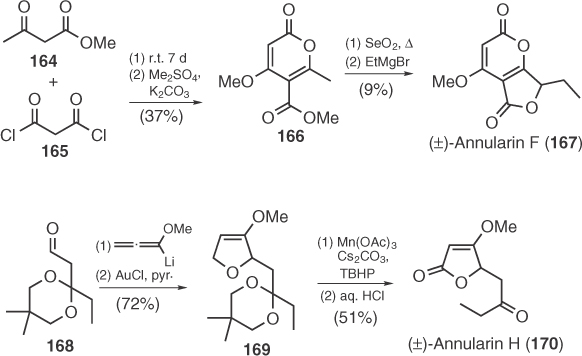
In 2010, Kato et al. [99] disclosed an enantioselective synthesis of annularin G and H, based on a Box-Pd(II)-catalyzed methoxycarbonylation of diastereoisomeric propargylic alcohols 171 and 173 (Scheme 1.26).
Scheme 1.26 Enantioselective synthesis of annularins G and H.
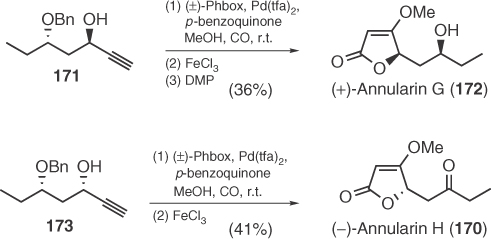
Palinurin (176), a linear furanosesterpene isolated from Ircinia sponges with anti-inflammatory and antibacterial properties [100], has been synthesized in 2009 by Fall's group [101]. The key step for the construction of the chiral tetronic acid moiety relies on the use of chiral pyrrolidine 174 (Scheme 1.27).
Scheme 1.27 Total synthesis of palinurin.
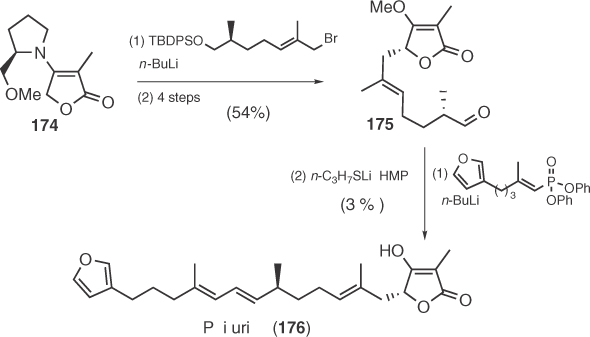
Pesthetoxin (180), a leaf necrosis inducing metabolite of the gray blight fungus Pestalotiopsis theae regularly infecting tea crops [102], was synthesized in a racemic form by Schobert et al. [103] in 2006 via the successive condensation of 177 with 2 equiv. of Ph3P=C=C=O and subsequent hydrolysis (Scheme 1.28).
Scheme 1.28 Synthesis of racemic pesthetoxin.
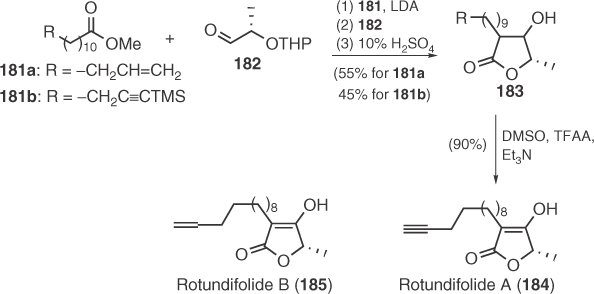
Rotundifolides A (184) and B (185), isolated from the bark of Litsea rotundifolia var. oblongifolia, were synthesized by the same group by a condensation of lactaldehyde 182 with suitable ester enolates and subsequent oxidation [104] (Scheme 1.29).
Scheme 1.29 Total synthesis of rotundifolides A and B.
(−)-Vertinolide (188), a “vertinoid” fungal metabolite isolated from Verticillium intertextum in 1982 by Dreiding et al., was first synthesized 1 year later by Ganem and Wrobel [105]. Special feature of their synthesis was the use of chiral sharpless epoxide 186 to provide vertinolide's precursor 187 after epoxide opening, lactonization, methylation, and ozonolysis (Scheme 1.30). The next year, Takaiwa and Yamashita [106] presented an alternative protocol based on homologation of 189 with the anion of methyl propanoate, followed by hydrolysis and relactonization to assemble vertinolide's core structure 190 (Scheme 1.30. In 1992, Schmidt et al. [107] synthesized Takaiwa's intermediate 190 by application of a stereoselective version of his reaction between β-alkoxy vinyl carbanions and ketones toward tetronic acids.
Scheme 1.30 Vertinolide syntheses by Wrobel and Takaiwa.
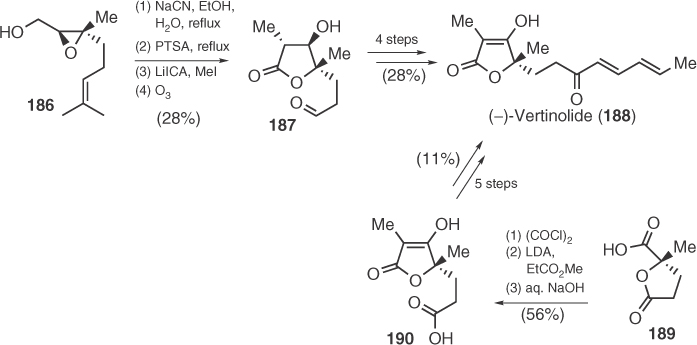
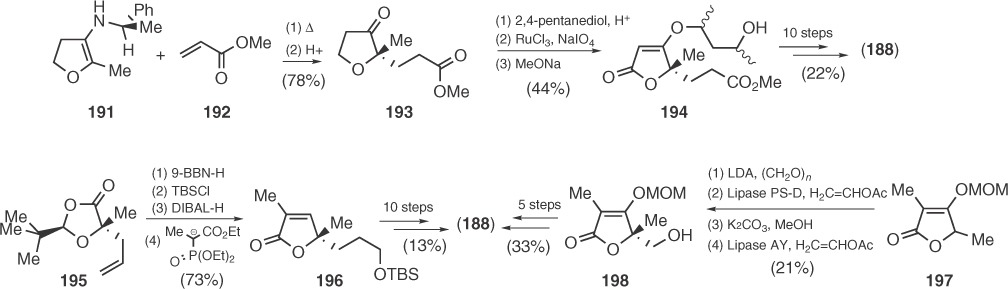
In 1992, Desmaële utilized enamine chemistry in a Michael addition to obtain chiral precursor 193 that gave vertinolide's tetronic ring (194) after ketalization, oxidation to lactone, and elimination (Scheme 1.31) [108]. After 4 years, Matsuo and Sakaguchi [109] employed their chiral 1,3-dioxolan-4-one 195 to obtain the central unsaturated lactone 196 after side-chain functionalization and condensation with triethyl-2-phosphonopropionate derived anion. Finally, in 2006, Takabe and coworkers [110], based on their previous work on the synthesis of racemic vertinolide, applied a lipase-catalyzed resolution on a 5-hydroxymethylene derivative of 197 to reach the natural enantiomer after five more synthetic steps (Scheme 1.31).
Scheme 1.31 Synthetic routes toward vertinolide.
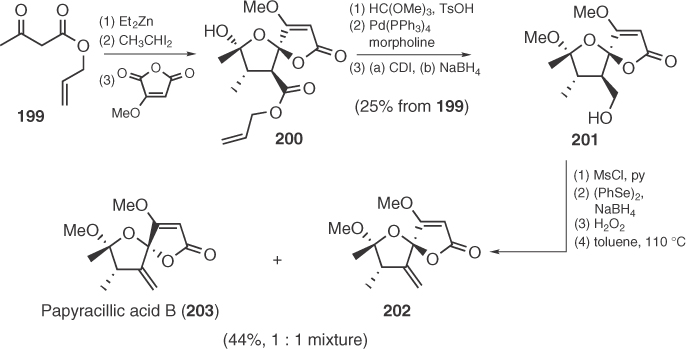
Papyracillic acid B (203), isolated from an endophytic fungus Microsphaeropsis sp., is a penicillic acid analog containing a spirofused cyclic ketal core appended with an exocyclic alkene [111]. Very recently, its racemic synthesis has been achieved by Zercher et al. [112] via a Zn-carbenoid-mediated chain extension–acylation reaction (Scheme 1.32). At the last step of Zercher's synthesis, paparycillic acid is obtained as 1 : 1 epimeric mixture with its C4 epimer, probably through an epimerization facilitated by exocyclic methylene group.
Scheme 1.32 Total synthesis of papyracillic acid B.
Bisorbibutenolide (or bislongiquinolide) (211), a member of sorbicilin-related natural products, is a biosynthetic product of bisorbicillinol (209), as it was reported in 1999 by Nicolaou et al. [113] in his inspiring work on this class of compounds. On the basis of biosynthetic considerations, Nicolaou converted 209 to 211 on treatment with potassium hexamethyldisilazide (KHMDS) in 80% yield (Scheme 1.33). Later in 2005, Deng et al. [114] prepared optically pure bisorbibutenolide by controlling the stereochemistry of sorbicillinol (207) starting from a cinchona (DHQ)2AQN-catalyzed cyanosilylation of 204–205.
Scheme 1.33 Total synthesis of bisorbibutenolide.

During the past three decades, a large amount of synthetic effort has been dedicated to studies on the spirotetronic antibiotics shown in Figure 1.2. These molecules share many structural similarities; however, some notable differences can also be detected. For example, all of them are 3-acyl tetronic derivatives with the exception of chlorothricolide (212) that bears an oxocarbonyl group at the same position. They all possess a macrocycle of different sizes spanning from the 13-membered ring in tetronolide (215) and kijanolide (216) to the impressive 32-membered ring of tetrameric quartromicins (219–220).
Figure 1.2 Spirotetronic antibiotics.
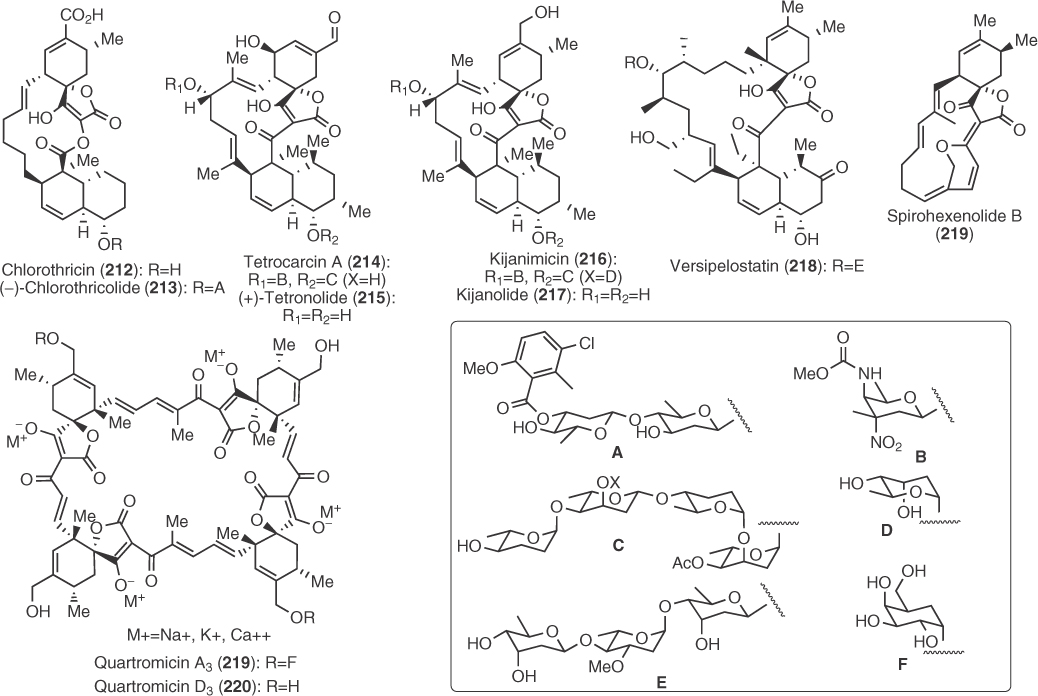
Synthetic studies mainly concern chlorothricolide, kijanolide, tetronolide, and quartromicins, while limited work has been done for versipelostatin (218) and spirohexenolide (219), the newer members of this family. Synthetic efforts have culminated in one total [115] and two analog syntheses [116, 117] of chlorothricolide, two total [118, 119] and one formal synthesis of tetronolide [120], one synthesis of a kijanolide analog [121] as well as numerous model or fragment syntheses on chlorothricolide [122–127], tetronolide [128–130], kijanolide [131–133], quartromicins [134, 135], versipelostatin [136], and spirohexenolide [137]. The discussion that follows will be limited only in the total and advanced analog syntheses mentioned earlier.
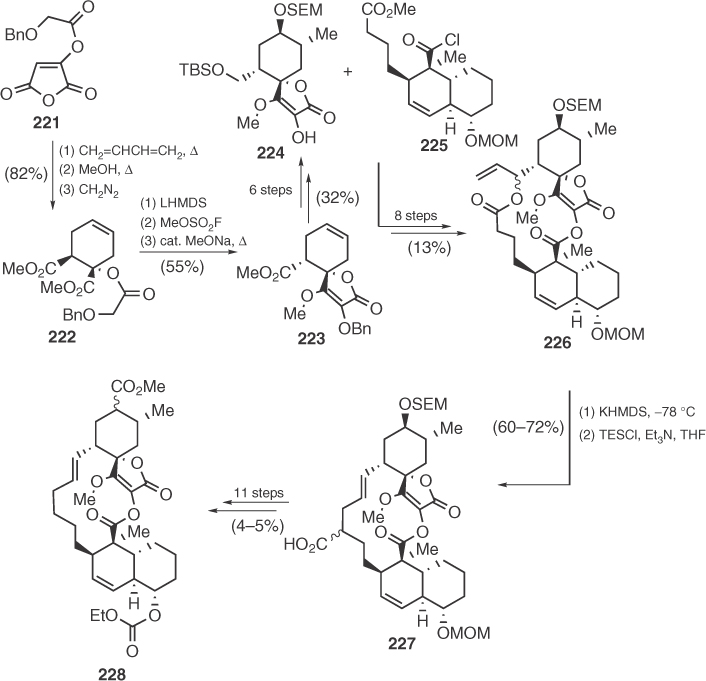
The first synthesis of an advanced analog of a spirotetronic antibiotic (228) was presented in 1986 by Ireland and Varney [116]. Ireland reached chlorothricolide's spirotetronic part via a route based on a Diels–Alder cycloaddition of maleic anhydride derivative 221 with 1,3-butadiene and a subsequent Dieckmann cyclization (Scheme 1.34). Coupling of the two chlorothricolide fragments 224 and 225 led to allyl carboxylate 226 that afforded the desired carbon macrocycle after a highly efficient Ireland–Claisen rearrangement. Transformation of 227 including an impressive radical decarboxylation led to the synthesis of protected dihydrochlorothricolide 228.
Scheme 1.34 Ireland's synthesis of chlorothricolide's analog 228.
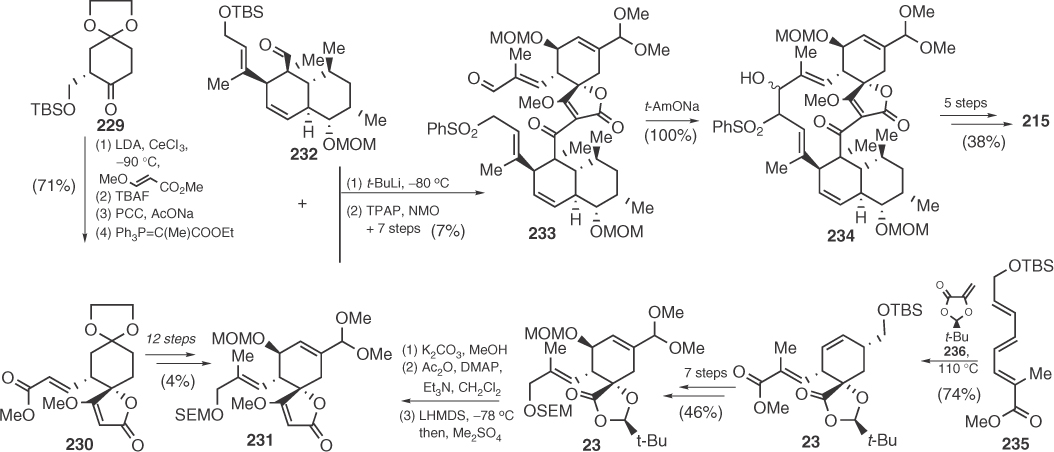
Significant advances in the synthesis of spirotetronic antibiotics were achieved during the following years because of the fundamental contribution of Yoshii and coworkers. In this context, in 1991, Yoshii et al. [118] presented the first total synthesis of a spirotetronic antibiotic, namely (+)-tetronolide (215). Yoshii performed an enantioselective synthesis of spirotetronic fragment 231 [128e] inspired by previous work of Schmidt on the reactions of vinyl carbanions using organocerium derivatives (Scheme 1.35) [126a]. Coupling of 231 with the octalin fragment 232 (prepared also enantioselectively by a Diels–Alder-based strategy) via an aldol type reaction furnished α-sulfonyl-ω-aldehyde 233 [128a], which was subjected to an elegant base-catalyzed macrocyclization [128c] for the assembly of the target macrocycle. It should be noted that, in 1997, Roush reported alternative strategies for the enantioselective synthesis of Yoshii's tetronolide fragments [120a, 129b], thus accomplishing a formal synthesis of this molecule. Interestingly, for the spirotetronic part 231, Roush employed an unexpectedly exo-diastereofacial selective Diels–Alder reaction between chiral dienophile 236 and triene 235.
Scheme 1.35 Yoshii's synthesis of (+)-tetronolide 215.
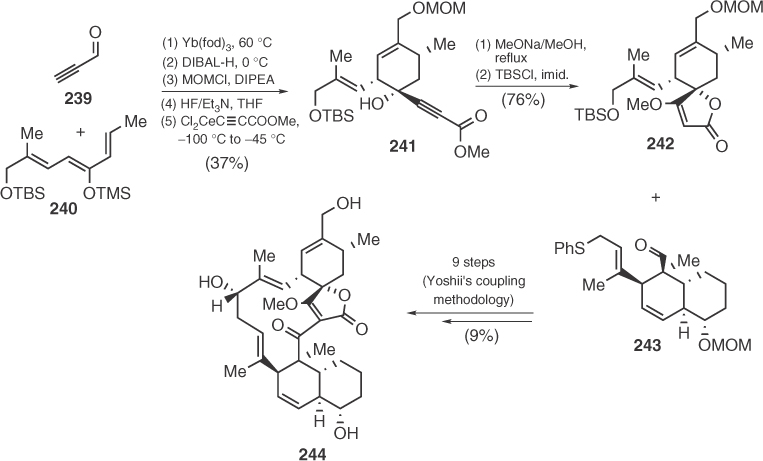
Although no total synthesis of kijanolide has ever been achieved, Yoshii managed to reach analog 26-O-methyl-28,29-bisnor-(±)-kijanolide (244) 3 years before the synthesis of tetronolide [121]. For the spirotetronic part, Yoshii applied Schmidt's chemistry by performing a reaction between a β,γ-unsaturated cyclohexanone and Cl2CeC≡CCOOMe followed by the base-catalyzed conversion of the resulting acetylenic carbinol 241 to spirotetronate 242 (Scheme 1.36).
Scheme 1.36 Yoshii's synthesis of kijanolide's analog 244.
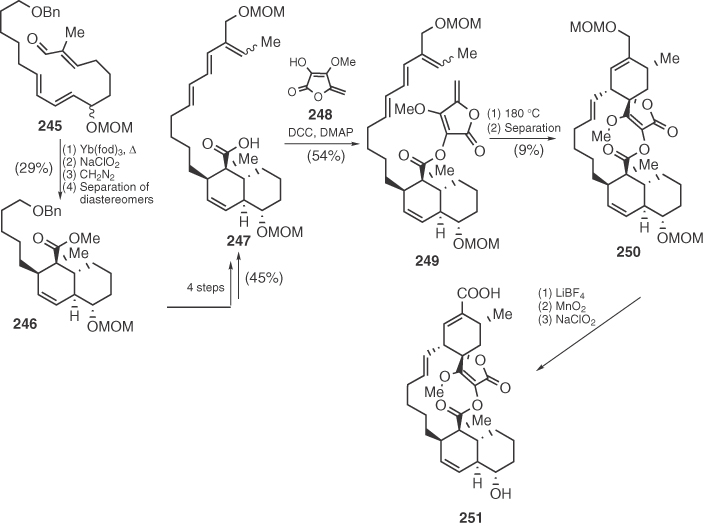
In addition, soon after Ireland's synthesis of chlorothricolide analog 228, Yoshii and coworkers [117] made one step further by presenting the synthesis of (±)-chlorothricolide as its 24-O-methyl derivative 251 (Scheme 1.37). Yoshii succeeded in utilizing the same strategy as in the total synthesis of ircinianin for the key step [138], that is, an intramolecular Diels–Alder reaction (IMDA) of intermediate 249 bearing an appropriate diene and a methylene tetronate. Despite the poor diastereofacial selectivity of this step, and the low overall efficiency, Yoshii's inventive strategy remains a masterful application of stereoselection via intramolecular cyclization.
Scheme 1.37 Yoshii's synthesis of chlorothricolide's analog 251.
After 13 years of numerous synthetic studies, Roush's group presented, in 1998, [115b] a brilliant enantioselective approach for the first total synthesis of (−)-chlorothricolide (Scheme 1.38). Roush designed a masterful strategy to build in one step the carbocyclic part of chlorothricolide, installing simultaneously seven stereocenters in excellent stereo- and enantioselectivity and high yield (252 → 253). This step was the result of intensive investigations on the relative rates of various Diels–Alder inter- and intracyclizations and is based on the use of a novel chiral dienophile (236) [123e,g] and the asymmetric inducing effect of various substituents on the polyene precursor 252 [123f].
Scheme 1.38 Roush's total synthesis of (−)-chlorothricolide.
